Variación genética humana
Puede haber múltiples variantes de cualquier gen en la población humana (alelos), una situación llamada polimorfismo.
Incluso los gemelos monocigóticos (que se desarrollan a partir de un cigoto) tienen diferencias genéticas poco frecuentes debido a las mutaciones que ocurren durante el desarrollo y la variación del número de copias del gen.
Las diferencias entre las poblaciones representan una pequeña proporción de la variación genética humana general.
El estudio de la variación genética humana tiene importancia evolutiva y aplicaciones médicas.
Es probable que los alelos bajo selección ocurran solo en aquellas regiones geográficas donde confieren una ventaja.
Los efectos fundadores seriales y el pequeño tamaño de la población anterior (que aumenta la probabilidad de deriva genética) pueden haber tenido una influencia importante en las diferencias neutrales entre las poblaciones.
Parece que un número pequeño pero significativo de genes se ha sometido a una selección natural reciente, y estas presiones selectivas a veces son específicas de una región.
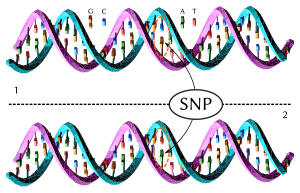
[12] A partir de 2017, la Single Nucleotide Polymorphism Database (DbSNP), que enumera SNP y otras variantes, enumeró 324 millones de variantes encontradas en genomas humanos secuenciados.
[14] Los SNP ocurren en promedio cada 100 a 300 bases[15] y, por lo tanto, son la principal fuente de heterogeneidad.
Un SNP funcional, o no sinónimo, es aquel que afecta algún factor, como el empalme de genes o el ARN mensajero, y por lo tanto causa una diferencia fenotípica entre los miembros de la especie.
Alrededor del 3% al 5% de los SNP humanos son funcionales (ver Proyecto Internacional HapMap).
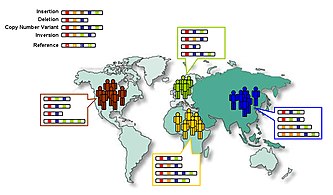
[18][19][20][21] Las variaciones del número de copias se heredan, pero también pueden surgir durante el desarrollo.
[30][31] En el estudio científico de la variación genética humana, un gen cline puede definirse rigurosamente y someterse a métricas cuantitativas.
Cada variante actúa como un alelo heredado, por lo que se utilizan para la identificación personal o parental.
[39] Se supone comúnmente que los primeros humanos abandonaron África y, por lo tanto, deben haber atravesado un cuello de botella en la población antes de su divergencia entre África y Eurasia hace unos 100,000 años (aproximadamente 3,000 generaciones).
En segundo lugar, los nuevos polimorfismos que surgieron en un grupo tenían menos probabilidades de transmitirse a otros grupos debido a que el flujo de genes estaba restringido.
El África subsahariana tiene la mayor diversidad genética humana y lo mismo se ha demostrado que es válido para la variación fenotípica en la forma del cráneo.
[35][43] El fenotipo está conectado al genotipo a través de la expresión génica.
La piel más oscura parece estar fuertemente seleccionada en regiones ecuatoriales para prevenir las quemaduras solares, el cáncer de piel, la fotólisis del folato y el daño a las glándulas sudoríparas.
[57] Se ha expresado que el FST no debe usarse como un marcador del estado de subespecies, ya que la estadística se usa para medir el grado de diferenciación entre poblaciones.
[60] Se ha criticado la aplicación de FST a poblaciones humanas, encontrando que la cifra del 85% es engañosa porque implica que todas las poblaciones humanas contienen en promedio el 85% de toda la diversidad genética.
Argumentan que el modelo estadístico subyacente asume incorrectamente historias de variación iguales e independientes para cada gran población humana.
[61] Existe la hipótesis de que los humanos anatómicamente modernos se cruzaron con los neandertales durante el Paleolítico Medio.
[10]Sin embargo, la identificación por origen geográfico puede romperse rápidamente cuando se considera la ascendencia histórica compartida entre individuos en el tiempo.
[71] Según algunos estudios, los métodos de prueba individuales, como las mediciones de la mitad del rostro y los rasgos del fémur, pueden identificar la ascendencia geográfica y, por extensión, la categoría racial a la que se habría asignado a un individuo durante su vida, con más del 80% de precisión y en combinación puede ser incluso más precisa.
[72] Las poblaciones de mezclas recientes que rastrean su ascendencia a múltiples continentes son muy adecuadas para identificar genes de rasgos y enfermedades que difieren en la prevalencia entre las poblaciones parentales.
[77] Incluso con enfermedades comunes que involucran numerosas variantes genéticas y factores ambientales, los investigadores señalan evidencia que sugiere la participación de alelos distribuidos diferencialmente con efectos pequeños a moderados.
Los ejemplos citados con frecuencia incluyen hipertensión, diabetes, obesidad y cáncer de próstata.
Algunas otras variaciones, por otro lado, son beneficiosas para los humanos, ya que previenen ciertas enfermedades y aumentan la posibilidad de adaptarse al medio ambiente.
[81] Sin embargo, en 2018 Noah Rosenberg publicó un estudio que argumentaba en contra de las ideas genéticamente esencialistas de las disparidades de salud entre las poblaciones, afirmando que las variantes ambientales son una causa más probable Interpretación de puntajes poligénicos, adaptación poligénica y diferencias fenotípicas humanas Los proyectos del genoma humano son esfuerzos científicos que determinan o estudian la estructura del genoma humano.