Los quistes pulmonares son igualmente frecuentes (84%) y el 24% de las personas con BDH acaban sufriendo un colapso pulmonar (neumotórax espontáneo).Aunque su función no se conoce del todo, parece ser un gen supresor de tumores que restringe el crecimiento y la división celular.Se han encontrado versiones del FLCN en otros animales, como moscas de la fruta, pastores alemanes, ratas y ratones.[5] Los tumores difieren entre individuos; pueden aparecer fusionados en placas, tener un aspecto similar a un comedón con un tapón de queratina o incluir quistes epidermoides.[6] Junto con los tumores, se observan otras afecciones cutáneas en las personas con síndrome de Birt-Hogg-Dubé.Estas lesiones suelen encontrarse en la axila, en los párpados y en los pliegues de la piel.Tanto los tumores benignos como los cancerosos pueden reducir la función renal con el tiempo a medida que aumentan de tamaño.[8] Junto con los fibrofoliculomas y los tumores renales, los individuos afectados desarrollan con frecuencia quistes (ampollas o bullas) en la base subpleural del pulmón o en el espacio intraparenquimatoso que pueden romperse y causar una acumulación anormal de aire en la cavidad torácica (neumotórax), que podría provocar el colapso de un pulmón.[6] Más del 83% de las personas con DHB tienen quistes, pero el síndrome no causa afecciones como la enfermedad pulmonar obstructiva crónica progresiva o la insuficiencia respiratoria generalizada,[2] aunque sí enfisema.[2] Se han observado neumotórax en personas de tan sólo 7 y 16 años.[2] Sin embargo, no se ha corroborado una conexión entre el síndrome BHD y el cáncer de tiroides.No se han encontrado diferencias significativas en los síntomas experimentados por las familias con una inserción en esa localización en comparación con las que tienen una deleción, pero las mutaciones en FLCN asociadas al síndrome de BHD son heterogéneas, y a menudo son mutaciones sin sentido o mutaciones por desplazamiento del marco de lectura (frameshift) que causan un truncamiento temprano del producto proteico en el extremo carboxi.[5] No se ha descubierto una correlación entre los distintos genotipos y fenotipos de FLCN.[5] También se ha encontrado en la glándula parótida, el cerebro, la mama, el páncreas, la próstata y los ovarios.[8] El extremo C-terminal de la foliculina ha demostrado ser el dominio a través del cual interactúa con FNIP1, y por lo tanto posiblemente con la vía mTOR.[14] Las personas con DHB nacen con una copia mutada del gen FLCN en cada célula.[6] La haploinsuficiencia -sólo tener una copia funcional del gen FLCN- es suficiente para causar los fibrofoliculomas y los quistes pulmonares, aunque una copia del gen es suficiente para mantener las células renales bajo control.Esta pérdida de heterocigosidad es un mecanismo común en el cáncer, y se detecta con frecuencia en los cánceres renales asociados a la BHD.[7] Se ha demostrado que la cistogénesis renal y la tumorigénesis en la BHD están impulsadas por la activación constitutiva de TFEB.[18] El BHD puede sugerirse por hallazgos clínicos, pero se diagnostica definitivamente mediante pruebas genéticas moleculares para detectar mutaciones en el gen FLCN.[5] Las manifestaciones cutáneas de la BHD se describieron originalmente como fibrofoliculomas (crecimientos anormales de un folículo piloso), tricodiscomas (lesiones hamartomatosas con un folículo piloso en la periferia, a menudo en la cara) y acrocordones (papilomas cutáneos).[9] Aunque los fibrofoliculomas son exclusivos del BHD, pueden presentar un aspecto ambiguo y deben confirmarse histológicamente.[6] Las distintas manifestaciones del BHD se controlan de diferentes maneras.Los fibrofoliculomas pueden extirparse quirúrgicamente, mediante curetaje, escisión por afeitado, rejuvenecimiento cutáneo o ablación con láser; sin embargo, ésta no es una solución permanente, ya que los tumores suelen reaparecer.Se considera que está infradiagnosticado[3][6] debido a la variabilidad de su expresión.El síndrome fue bien descrito por primera vez en 1977,[21] por tres médicos canadienses, Arthur R. Birt, Georgina R. Hogg y William J. Dubé.[23] Aunque los hermanos no presentaban síntomas renales ni pulmonares, su padre tenía quistes en los pulmones y los riñones.[3] El síndrome de Hornstein-Knickenberg es un nombre ahora en desuso para los fibrofoliculomas hereditarios inherentes a la EHB.[6] Las hembras de pastor alemán con una mutación FLCN también son propensas a padecer leiomiomas uterinos.Presentan una mutación en el homólogo de FLCN que produce una proteína truncada, aunque no desarrollan los síntomas cutáneos o pulmonares observados en humanos.
Tinción H&E
de tejido de un carcinoma cromófobo de células renales, el segundo cáncer más común asociado al BHD.
Este diagrama muestra cómo se transmiten los trastornos autosómicos dominantes como el BHD. El progenitor no afectado produce todos los gametos normales (espermatozoides y óvulos) y el progenitor afectado produce la mitad de gametos mutantes y la otra mitad de gametos normales. Dado que sólo se necesita una copia de la mutación para tener una enfermedad autosómica dominante, cada descendiente tiene un 50% de probabilidades de tener la mutación.
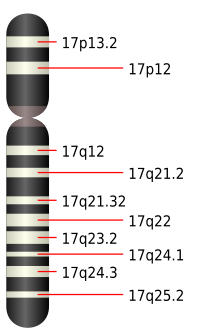
La foliculina está codificada por el gen FLCN, situado en el
brazo p
del cromosoma 17 humano.
La persona de esta imagen padece esclerosis tuberosa. Las lesiones cutáneas causadas por la
esclerosis tuberosa
(angiofibromas) deben distinguirse de los fibrofoliculomas característicos de la EHB, que también se producen principalmente en la cara.