Síndrome de West
El síndrome de West (SW) o síndrome de los espasmos infantiles es una encefalopatía (alteración cerebral) epiléptica de la infancia, grave y poco frecuente, que debe su nombre a William James West (1793-1848), médico inglés que describió por primera vez el cuadro (presente en su propio hijo) en un artículo publicado por The Lancet en 1841.
Los niños con SW suelen manifestar la enfermedad entre los 3 y 6 meses de edad, aunque en ocasiones esto ocurre hasta los dos años.
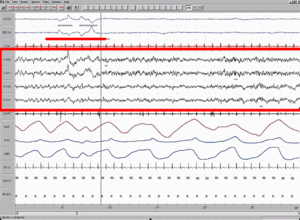
Diversos autores[7] postulan que un desequilibrio en la producción de neurotransmisores del tallo cerebral podría originar la hipsarritmia y los espasmos epilépticos (por aumento de los sistemas serotoninérgico o adrenérgico, o por inhibición del sistema colinérgico).
[8][9] No existe una clara asociación familiar (excepto en la variedad relacionada con la esclerosis tuberosa), ni siquiera con otros cuadros epilépticos.
Con menos frecuencia aparecen en los primeros dos meses de vida, o entre los dos y los cuatro años.
Los espasmos rara vez se presentan aislados: suelen ocurrir en salvas (típicamente al despertarse, o antes de dormirse) y son muy poco frecuentes durante el sueño.
Otros cuadros con los que se puede confundir son: Este síndrome tiene, en general, mal pronóstico.
Son de mejor pronóstico las variedades idiopáticas o criptogenéticas, y peor en los casos más sintomáticos.
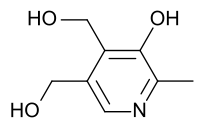
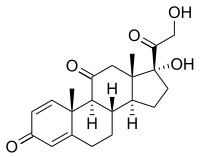
Suele ser una técnica eficaz en la resolución de las crisis, aunque su eficacia en el desarrollo psicomotor es más controvertida.