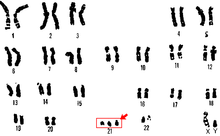
Síndrome de Down
Se caracteriza por un grado variable de discapacidad intelectual y unos rasgos físicos que le dan un aspecto reconocible.
Los avances actuales en el descifrado del genoma humano están revelando algunos de los procesos bioquímicos subyacentes a la discapacidad intelectual, pero en la actualidad no existe ningún tratamiento farmacológico que haya demostrado que mejora las capacidades intelectuales de estas personas.
P. Martin Duncan en 1886 describe textualmente a «una niña de cabeza pequeña, redondeada, con ojos achinados, que dejaba colgar la lengua y apenas pronunciaba unas pocas palabras».
Tras varias comunicaciones científicas, finalmente en 1909 G. E. Shuttleworth mencionó por primera vez la edad materna avanzada como un factor de riesgo para la aparición del síndrome.
[14] De camino a la denominación el síndrome fue rebautizado como «idiocia Kalmukia» o «niños inconclusos».
Algún autor[21] también ha relacionado la baja frecuencia coital así como el uso de anovulatorios y espermicidas con la aparición del síndrome.
Esto hay que ponderarlo para cada caso con el riesgo propio de la madre según su edad.
Este hecho permitió estudiar la variabilidad genómica debida al síndrome de Down eliminando la variación genética entre muestras.
Estos dominios de expresión génica desregulada se conocen como GEDDs (Gene expression dysregulation domains).
La organización en dominios en los gemelos discordantes podría ser atribuida fundamentalmente a la copia extra del cromosoma 21.
Estas regiones presentan una baja densidad de genes y una expresión génica disminuida entre otras características.
Esto es interesante porque la perturbación de la regulación génica puede ser común a otras anomalías cromosómicas.
Como rasgos comunes se pueden reseñar su fisionomía peculiar, una hipotonía muscular generalizada, un grado variable de discapacidad cognitiva y retardo en el crecimiento.
Una tercera parte (en torno al 30 % según las fuentes) son defectos de cierre del septo ventricular (pared que separa los ventrículos entre sí), y con menos frecuencia se encuentran otras enfermedades como ostium secundum,[35] ductus arterioso persistente[36] o tetralogía de Fallot.
[37] En general casi todos estos defectos provocan paso inapropiado de sangre desde las cavidades izquierdas del corazón a las derechas, lo que aumenta la circulación pulmonar.
Esta es una patología grave que precisa cirugía, habitualmente en el primer año de vida, para reparar los defectos.
Por este motivo se recomienda la realización de una ecografía del corazón a todo recién nacido con SD.
[40] El ano imperforado es la malformación anorrectal más frecuente en niños con SD: se ha descrito una incidencia del 2-3 %[41] (es decir, dos o tres da cada cien niños recién nacidos con SD lo presentan), mientras que su aparición en la población general se estima en torno a uno de cada 5000.
Otros trastornos relativamente frecuentes son el megacolon, o dilatación excesiva de la porción distal del tracto digestivo por un defecto en la relajación y la enfermedad celíaca (intolerancia digestiva al gluten), que aparecen también con una frecuencia superior a la que se presenta en recién nacidos sin el síndrome.
Suele tratarse de hipotiroidismos leves adquiridos o autoinmunes que en muchos casos no precisan tratamiento, aunque cuando su gravedad lo requiere deben instaurarse lo más precozmente posible para no ver comprometido el potencial de desarrollo intelectual.
Las personas con SD presentan ciertas anomalías inmunológicas de características e intensidad variables que no son fácilmente asimilables a las inmunodeficiencias catalogadas.
Es una técnica relativamente inocua y poco molesta, pero comporta un riesgo del 1-2 % de aborto, lesión fetal, o infección materna.
A lo largo de los últimos 150 años se han postulado diferentes tratamientos empíricos (hormona tiroidea, hormona del crecimiento, ácido glutámico, dimetilsulfóxido, complejos vitamínicos y minerales, 5-Hidroxitriptófano o piracetam) sin que ninguno haya demostrado en estudios longitudinales a doble ciego que su administración provoque ningún efecto positivo significativo en el desarrollo motor, social, intelectual o de expresión verbal de las personas con SD.
No existe hasta la fecha ningún tratamiento farmacológico eficaz para el SD, aunque los estudios puestos en marcha con la secuenciación del genoma humano permiten augurar una posible vía de actuación (enzimática o genética), eso sí, en un futuro todavía algo lejano.
[44][45] Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial.
La adaptación curricular permite en muchos casos una integración normalizada en colegios habituales, aunque deben tenerse en cuenta sus necesidades educativas especiales.
Como consecuencia, es imposible determinar los trabajos y desempeños que pueden conseguir durante la vida adulta.
Dicho compuesto afecta al gen DYRK1A, relacionado con la formación del cerebro y sobreactivado por el cromosoma extra del síndrome de Down; este gen produce un exceso de proteínas asociadas a las alteraciones cognitivas que este compuesto devuelve a sus niveles normales.
[47] Además el objetivo de estos programas no es tan solo la adquisición de habilidades, sino que estas se alcancen mucho antes, permitiendo continuar con programas educativos que integren al máximo a la persona con SD en entornos normalizados.
[42][49] Se desconocen todavía los mecanismos que provocan la discapacidad en las personas con SD, aunque la secuenciación del genoma humano y diversos estudios llevados a cabo en sujetos con translocaciones parciales están empezando a servir para descubrir los genes responsables del cuadro.