
La química computacional es una rama de la química que utiliza simulaciones por computadora para ayudar a resolver problemas químicos. [2] Utiliza métodos de química teórica incorporados a programas informáticos para calcular las estructuras y propiedades de moléculas , grupos de moléculas y sólidos. [3] La importancia de este tema surge del hecho de que, con la excepción de algunos hallazgos relativamente recientes relacionados con el ion molecular de hidrógeno ( catión dihidrógeno ), lograr una representación mecánica cuántica precisa de los sistemas químicos de forma analítica, o en forma cerrada, no es factible. [4] La complejidad inherente al problema de muchos cuerpos exacerba el desafío de proporcionar descripciones detalladas de los sistemas mecánicos cuánticos. [5] Si bien los resultados computacionales normalmente complementan la información obtenida mediante experimentos químicos , ocasionalmente pueden predecir fenómenos químicos no observados . [6]
La química computacional se diferencia de la química teórica , que implica una descripción matemática de la química. Sin embargo, la química computacional implica el uso de programas informáticos y habilidades matemáticas adicionales para modelar con precisión varios problemas químicos. En química teórica, los químicos, físicos y matemáticos desarrollan algoritmos y programas informáticos para predecir propiedades atómicas y moleculares y rutas de reacción para reacciones químicas. Los químicos computacionales, por el contrario, pueden simplemente aplicar programas y metodologías informáticas existentes a cuestiones químicas específicas. [7]
Históricamente, la química computacional ha tenido dos vertientes diferenciadas:
Estos aspectos, junto con el propósito de la química computacional, han dado como resultado una gran cantidad de algoritmos.
Partiendo de los descubrimientos y teorías fundacionales de la historia de la mecánica cuántica , los primeros cálculos teóricos en química fueron los de Walter Heitler y Fritz London en 1927, utilizando la teoría del enlace de valencia . [9] Los libros que influyeron en el desarrollo inicial de la química cuántica computacional incluyen Introducción a la mecánica cuántica de 1935 de Linus Pauling y E. Bright Wilson , con aplicaciones a la química , [10] Química cuántica de 1944 de Eyring , Walter y Kimball , [ 11] Mecánica de ondas elementales de 1945 de Heitler : con aplicaciones a la química cuántica , [12] y más tarde el libro de texto Valence de Coulson de 1952 , cada uno de los cuales sirvió como referencia principal para los químicos en las décadas siguientes. [13]
Con el desarrollo de tecnología informática eficiente en la década de 1940, las soluciones de elaboradas ecuaciones de onda para sistemas atómicos complejos comenzaron a ser un objetivo realizable. A principios de la década de 1950 se realizaron los primeros cálculos semiempíricos de orbitales atómicos. Los químicos teóricos se convirtieron en usuarios extensivos de las primeras computadoras digitales. Un avance significativo lo marcó el artículo de Clemens CJ Roothaan de 1951 en Reviews of Modern Physics. [14] [15] Este artículo se centró en gran medida en el enfoque "LCAO MO" (Combinación lineal de orbitales atómicos y orbitales moleculares). Durante muchos años, fue el segundo artículo más citado en esa revista. [14] [15] Smith y Sutcliffe dan una descripción muy detallada de dicho uso en el Reino Unido. [16] Los primeros cálculos ab initio con el método Hartree-Fock sobre moléculas diatómicas se realizaron en 1956 en el MIT, utilizando un conjunto básico de orbitales de Slater . [17] Para las moléculas diatómicas, Ransil y Nesbet publicaron respectivamente en 1960 un estudio sistemático utilizando un conjunto de bases mínimo y el primer cálculo con un conjunto de bases más grande. [18] Los primeros cálculos poliatómicos utilizando orbitales gaussianos se realizaron a finales de la década de 1950 . Los primeros cálculos de interacción de configuración se realizaron en Cambridge en la computadora EDSAC en la década de 1950 utilizando orbitales gaussianos por Boys y sus compañeros. [19] En 1971, cuando se publicó una bibliografía de cálculos ab initio , [20] las moléculas más grandes incluidas fueron la naftaleno y el azuleno . [21] [22] Schaefer ha publicado resúmenes de muchos desarrollos anteriores en la teoría ab initio . [23]
En 1964 , se generaron en computadoras en Berkeley y Oxford. [24] Estos métodos empíricos fueron reemplazados en la década de 1960 por métodos semiempíricos como CNDO . [25]
A principios de la década de 1970, se empezaron a utilizar programas informáticos ab initio eficientes, como ATMOL, Gaussiano , IBMOL y POLYAYTOM, para acelerar los cálculos ab initio de orbitales moleculares. [26] De estos cuatro programas, sólo el gaussiano, ahora enormemente ampliado, sigue en uso, pero ahora se utilizan muchos otros programas. [26] Al mismo tiempo, los métodos de la mecánica molecular , como el campo de fuerza MM2 , fueron desarrollados, principalmente por Norman Allinger . [27]
Una de las primeras menciones del término química computacional se puede encontrar en el libro de 1970 Computers and Their Role in the Physical Sciences de Sidney Fernbach y Abraham Haskell Taub, donde afirman: "Parece, por tanto, que la 'química computacional' finalmente puede ser cada vez más una realidad." [28] Durante la década de 1970, métodos muy diferentes comenzaron a verse como parte de una nueva disciplina emergente de la química computacional . [29] El Journal of Computational Chemistry se publicó por primera vez en 1980.
La química computacional ha sido galardonada con varios premios Nobel, sobre todo en 1998 y 2013. Walter Kohn , "por su desarrollo de la teoría de la densidad funcional", y John Pople , "por su desarrollo de métodos computacionales en química cuántica", recibieron el premio. 1998 Premio Nobel de Química. [30] Martin Karplus , Michael Levitt y Arieh Warshel recibieron el Premio Nobel de Química 2013 por "el desarrollo de modelos multiescala para sistemas químicos complejos". [31]
Hay varios campos dentro de la química computacional.
Estos campos pueden dar lugar a varias aplicaciones como se muestra a continuación.

La química computacional es una herramienta para analizar sistemas catalíticos sin realizar experimentos. La teoría moderna de la estructura electrónica y la teoría funcional de la densidad han permitido a los investigadores descubrir y comprender los catalizadores . [37] Los estudios computacionales aplican la química teórica a la investigación de la catálisis. Los métodos de la teoría del funcional de la densidad calculan las energías y los orbitales de las moléculas para dar modelos de esas estructuras. [38] Utilizando estos métodos, los investigadores pueden predecir valores como la energía de activación , la reactividad del sitio [39] y otras propiedades termodinámicas. [38]
Los datos que son difíciles de obtener experimentalmente se pueden encontrar utilizando métodos computacionales para modelar los mecanismos de los ciclos catalíticos. [39] Los químicos computacionales expertos proporcionan predicciones que se acercan a los datos experimentales con consideraciones adecuadas de los métodos y conjuntos de bases. Con buenos datos computacionales, los investigadores pueden predecir cómo se pueden mejorar los catalizadores para reducir el costo y aumentar la eficiencia de estas reacciones. [38]
La química computacional se utiliza en el desarrollo de fármacos para modelar moléculas de fármacos potencialmente útiles y ayudar a las empresas a ahorrar tiempo y costos en el desarrollo de fármacos. El proceso de descubrimiento de fármacos implica analizar datos, encontrar formas de mejorar las moléculas actuales, encontrar rutas sintéticas y probar esas moléculas. [36] La química computacional ayuda con este proceso al dar predicciones sobre qué experimentos sería mejor realizar sin realizar otros experimentos. Los métodos computacionales también pueden encontrar valores que son difíciles de encontrar experimentalmente como el pKa de los compuestos. [40] Se pueden utilizar métodos como la teoría funcional de la densidad para modelar moléculas de fármacos y encontrar sus propiedades, como sus energías HOMO y LUMO y orbitales moleculares. Los químicos computacionales también ayudan a las empresas a desarrollar informática, infraestructura y diseños de medicamentos. [41]
Además de la síntesis de fármacos, los químicos computacionales también investigan los portadores de fármacos en busca de nanomateriales . Permite a los investigadores simular entornos para probar la eficacia y estabilidad de los portadores de fármacos. Comprender cómo interactúa el agua con estos nanomateriales garantiza la estabilidad del material en el cuerpo humano. Estas simulaciones computacionales ayudan a los investigadores a optimizar el material y encontrar la mejor manera de estructurar estos nanomateriales antes de fabricarlos. [42]
Las bases de datos son útiles para los químicos computacionales y no computacionales en la investigación y verificación de la validez de los métodos computacionales. Los datos empíricos se utilizan para analizar el error de los métodos computacionales frente a los datos experimentales. Los datos empíricos ayudan a los investigadores con sus métodos y conjuntos de bases a tener una mayor confianza en los resultados de los investigadores. Las bases de datos de química computacional también se utilizan para probar software o hardware para química computacional. [43]
Las bases de datos también pueden utilizar datos puramente calculados. Los datos puramente calculados utilizan valores calculados sobre valores experimentales para bases de datos. Los datos puramente calculados evitan tener que lidiar con estos ajustes para diferentes condiciones experimentales, como la energía de punto cero. Estos cálculos también pueden evitar errores experimentales en moléculas difíciles de probar. Aunque los datos puramente calculados a menudo no son perfectos, identificar problemas suele ser más fácil para los datos calculados que para los experimentales. [43]
Las bases de datos también brindan acceso público a la información para que la utilicen los investigadores. Contienen datos que otros investigadores han encontrado y cargado en estas bases de datos para que cualquiera pueda buscarlos. Los investigadores utilizan estas bases de datos para encontrar información sobre moléculas de interés y aprender qué se puede hacer con esas moléculas. Algunas bases de datos de química disponibles públicamente incluyen las siguientes. [43]
Los programas utilizados en química computacional se basan en muchos métodos químicos cuánticos diferentes que resuelven la ecuación molecular de Schrödinger asociada con el hamiltoniano molecular . [46] Los métodos que no incluyen ningún parámetro empírico o semiempírico en sus ecuaciones (derivados directamente de la teoría, sin inclusión de datos experimentales) se denominan métodos ab initio . [47] Una aproximación teórica se define rigurosamente sobre los primeros principios y luego se resuelve dentro de un margen de error que se conoce cualitativamente de antemano. Si se deben utilizar métodos numéricos iterativos, el objetivo es iterar hasta obtener la precisión total de la máquina (la mejor posible con una longitud de palabra finita en la computadora y dentro de las aproximaciones matemáticas y/o físicas realizadas). [48]
Los métodos ab initio necesitan definir un nivel de teoría (el método) y un conjunto de bases. [49] Un conjunto de bases consta de funciones centradas en los átomos de la molécula. Estos conjuntos luego se utilizan para describir orbitales moleculares mediante el método de orbitales moleculares de combinación lineal de orbitales atómicos (LCAO) ansatz . [50]

Un tipo común de cálculo ab initio de la estructura electrónica es el método Hartree-Fock (HF), una extensión de la teoría de los orbitales moleculares , donde las repulsiones electrón-electrón en la molécula no se tienen en cuenta específicamente; sólo el efecto promedio de los electrones se incluye en el cálculo. A medida que aumenta el tamaño del conjunto de bases, la energía y la función de onda tienden hacia un límite llamado límite de Hartree-Fock. [50]
Muchos tipos de cálculos comienzan con un cálculo de Hartree-Fock y posteriormente corrigen la repulsión electrón-electrón, también conocida como correlación electrónica . [51] Estos tipos de cálculos se denominan métodos post-Hartree-Fock . Mejorando continuamente estos métodos, los científicos pueden acercarse cada vez más a predecir perfectamente el comportamiento de los sistemas atómicos y moleculares en el marco de la mecánica cuántica, tal como la define la ecuación de Schrödinger. [52] Para obtener una concordancia exacta con el experimento, es necesario incluir términos específicos, algunos de los cuales son mucho más importantes para los átomos pesados que para los más ligeros. [53]
En la mayoría de los casos, la función de onda de Hartree-Fock ocupa una única configuración o determinante. [54] En algunos casos, particularmente para los procesos de ruptura de enlaces, esto es inadecuado y se deben usar varias configuraciones . [55]
La energía molecular total se puede evaluar en función de la geometría molecular ; en otras palabras, la superficie de energía potencial . [56] Una superficie de este tipo se puede utilizar para la dinámica de reacciones. Los puntos estacionarios de la superficie conducen a predicciones de diferentes isómeros y estructuras de transición para la conversión entre isómeros, pero éstas pueden determinarse sin un conocimiento completo de la superficie completa. [53]

Un objetivo particularmente importante, llamado termoquímica computacional , es calcular cantidades termoquímicas como la entalpía de formación con precisión química. La precisión química es la precisión necesaria para realizar predicciones químicas realistas y generalmente se considera 1 kcal/mol o 4 kJ/mol. Para alcanzar esa precisión de forma económica, es necesario utilizar una serie de métodos post-Hartree-Fock y combinar los resultados. Estos métodos se denominan métodos compuestos de química cuántica . [57]
Después de separar las variables electrónica y nuclear dentro de la representación de Born-Oppenheimer), el paquete de ondas correspondiente a los grados de libertad nucleares se propaga a través del operador de evolución temporal (física) asociado a la ecuación de Schrödinger dependiente del tiempo (para el hamiltoniano molecular completo). ). [58] En el enfoque complementario dependiente de la energía, la ecuación de Schrödinger independiente del tiempo se resuelve utilizando el formalismo de la teoría de la dispersión . El potencial que representa la interacción interatómica viene dado por las superficies de energía potencial . En general, las superficies de energía potencial se acoplan mediante términos de acoplamiento vibrónico . [59]
Los métodos más populares para propagar el paquete de ondas asociado a la geometría molecular son:
La forma en que un método computacional resuelve ecuaciones cuánticas afecta la precisión y eficiencia del método. La técnica del operador dividido es uno de estos métodos para resolver ecuaciones diferenciales. En química computacional, la técnica del operador dividido reduce los costos computacionales de la simulación de sistemas químicos. Los costos computacionales se refieren al tiempo que les toma a las computadoras calcular estos sistemas químicos, ya que los sistemas más complejos pueden tardar días. Los sistemas cuánticos son difíciles de resolver para los humanos y requieren mucho tiempo. Los métodos de operador dividido ayudan a las computadoras a calcular estos sistemas rápidamente resolviendo los subproblemas en una ecuación diferencial cuántica . El método hace esto separando la ecuación diferencial en dos ecuaciones diferentes, como cuando hay más de dos operadores. Una vez resueltas, las ecuaciones divididas se combinan nuevamente en una sola ecuación para dar una solución fácilmente calculable. [62]
Este método se utiliza en muchos campos que requieren resolver ecuaciones diferenciales, como la biología . Sin embargo, la técnica conlleva un error de división. Por ejemplo, con la siguiente solución para una ecuación diferencial. [62]
La ecuación se puede dividir, pero las soluciones no serán exactas, sólo similares. Este es un ejemplo de división de primer orden. [62]
Hay formas de reducir este error, que incluyen tomar un promedio de dos ecuaciones divididas. [62]
Otra forma de aumentar la precisión es utilizar una división de orden superior. Por lo general, la división de segundo orden es lo máximo que se hace porque la división de orden superior requiere mucho más tiempo para calcularse y no vale la pena el costo. Los métodos de orden superior se vuelven demasiado difíciles de implementar y no son útiles para resolver ecuaciones diferenciales a pesar de su mayor precisión. [62]
Los químicos computacionales dedican mucho tiempo a hacer que los sistemas calculados con la técnica del operador dividido sean más precisos y al mismo tiempo minimicen el costo computacional. Calcular métodos es un desafío enorme para muchos químicos que intentan simular moléculas o entornos químicos. [62]

Los métodos de la teoría funcional de la densidad (DFT) a menudo se consideran métodos ab initio para determinar la estructura electrónica molecular, aunque muchos de los funcionales más comunes utilizan parámetros derivados de datos empíricos o de cálculos más complejos. En DFT, la energía total se expresa en términos de la densidad total de un electrón en lugar de la función de onda. En este tipo de cálculo, hay un hamiltoniano aproximado y una expresión aproximada para la densidad electrónica total. Los métodos DFT pueden ser muy precisos con un coste computacional reducido. Algunos métodos combinan el intercambio funcional de densidad con el término de intercambio Hartree-Fock y se denominan métodos funcionales híbridos . [63]
Los métodos semiempíricos de la química cuántica se basan en el formalismo del método Hartree-Fock , pero realizan muchas aproximaciones y obtienen algunos parámetros a partir de datos empíricos. Fueron muy importantes en la química computacional desde los años 60 hasta los 90, especialmente para el tratamiento de moléculas grandes donde el método Hartree-Fock completo sin las aproximaciones era demasiado costoso. El uso de parámetros empíricos parece permitir cierta inclusión de efectos de correlación en los métodos. [64]
Los métodos semiempíricos primitivos se diseñaron incluso antes, donde la parte de dos electrones del hamiltoniano no está incluida explícitamente. Para los sistemas de electrones π, este fue el método de Hückel propuesto por Erich Hückel , y para todos los sistemas de electrones de valencia, el método de Hückel extendido propuesto por Roald Hoffmann . A veces, los métodos de Hückel se denominan "completamente empíricos" porque no se derivan de un hamiltoniano. [65] Sin embargo, el término "métodos empíricos" o "campos de fuerza empíricos" se utiliza habitualmente para describir la mecánica molecular. [66]
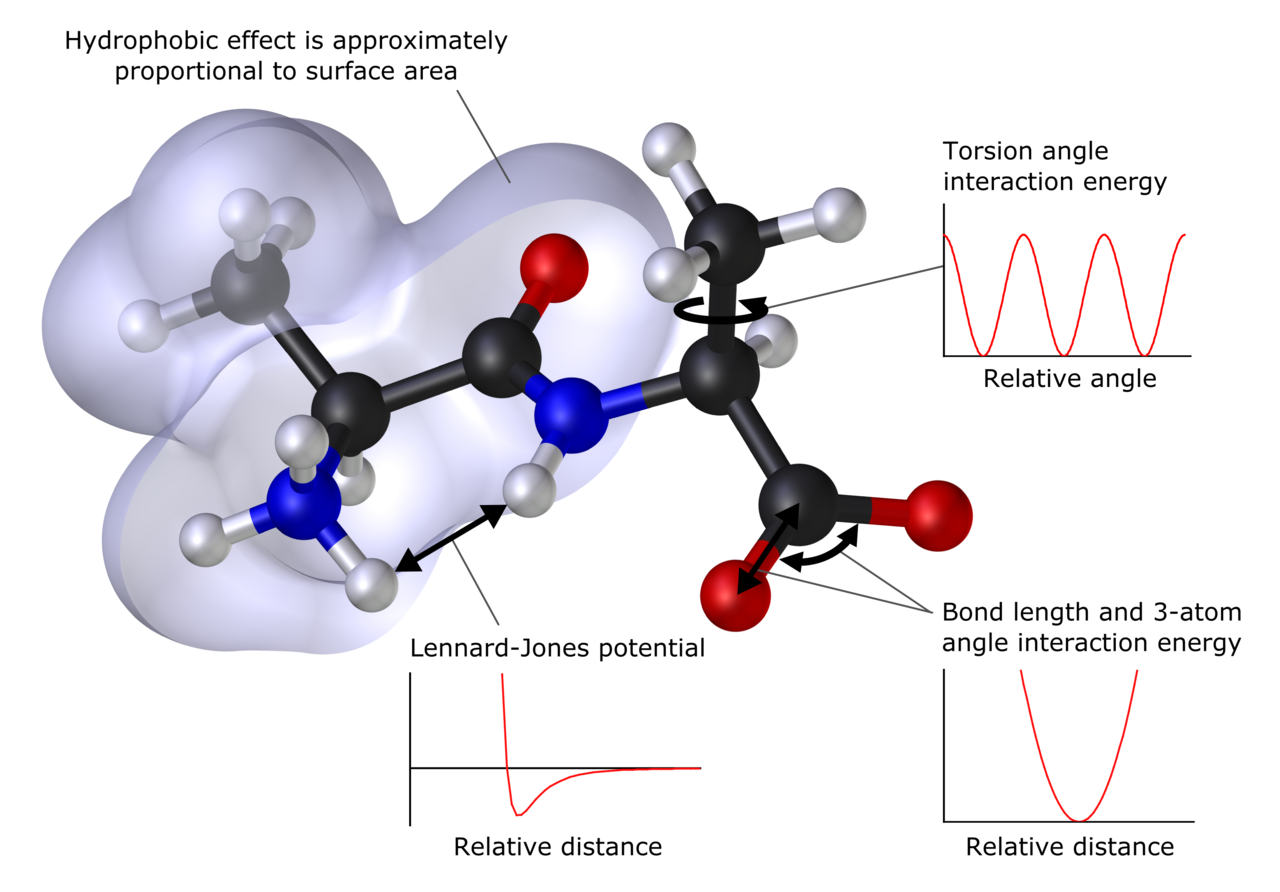
En muchos casos, se pueden modelar con éxito grandes sistemas moleculares evitando por completo los cálculos de la mecánica cuántica. Las simulaciones de mecánica molecular , por ejemplo, utilizan una expresión clásica para la energía de un compuesto, por ejemplo, el oscilador armónico . Todas las constantes que aparecen en las ecuaciones deben obtenerse previamente a partir de datos experimentales o cálculos ab initio . [64]
La base de datos de compuestos utilizados para la parametrización, es decir, el conjunto resultante de parámetros y funciones se denomina campo de fuerza , es crucial para el éxito de los cálculos de mecánica molecular. Se esperaría que un campo de fuerza parametrizado contra una clase específica de moléculas, por ejemplo, proteínas, sólo tuviera relevancia al describir otras moléculas de la misma clase. [64] Estos métodos se pueden aplicar a proteínas y otras moléculas biológicas grandes, y permiten estudios del acercamiento y la interacción (acoplamiento) de posibles moléculas de fármacos. [67] [68]
La dinámica molecular (DM) utiliza la mecánica cuántica , la mecánica molecular o una combinación de ambas para calcular fuerzas que luego se utilizan para resolver las leyes del movimiento de Newton para examinar el comportamiento de los sistemas dependiente del tiempo. El resultado de una simulación de dinámica molecular es una trayectoria que describe cómo la posición y la velocidad de las partículas varían con el tiempo. El punto de fase de un sistema descrito por las posiciones y los momentos de todas sus partículas en un punto de tiempo anterior determinará el siguiente punto de fase mediante la integración de las leyes de movimiento de Newton. [69]
Monte Carlo (MC) genera configuraciones de un sistema realizando cambios aleatorios en las posiciones de sus partículas, junto con sus orientaciones y conformaciones cuando corresponda. [70] Es un método de muestreo aleatorio, que hace uso del llamado muestreo de importancia . Los métodos de muestreo de importancia pueden generar estados de baja energía, ya que esto permite calcular las propiedades con precisión. La energía potencial de cada configuración del sistema se puede calcular, junto con los valores de otras propiedades, a partir de las posiciones de los átomos. [71] [72]
QM/MM es un método híbrido que intenta combinar la precisión de la mecánica cuántica con la velocidad de la mecánica molecular. Es útil para simular moléculas muy grandes, como enzimas . [73]
La química computacional cuántica tiene como objetivo explotar la computación cuántica para simular sistemas químicos, distinguiéndose del enfoque QM/MM (Mecánica Cuántica/Mecánica Molecular). [74] Mientras que QM/MM utiliza un enfoque híbrido, combinando la mecánica cuántica para una parte del sistema con la mecánica clásica para el resto, la química computacional cuántica utiliza exclusivamente métodos de computación cuántica para representar y procesar información, como los operadores hamiltonianos. [75]
Los métodos convencionales de química computacional a menudo tienen problemas con las complejas ecuaciones de la mecánica cuántica, particularmente debido al crecimiento exponencial de la función de onda de un sistema cuántico. La química computacional cuántica aborda estos desafíos utilizando métodos de computación cuántica , como la qubitización y la estimación de fase cuántica , que se cree que ofrecen soluciones escalables. [76]
La qubitización implica adaptar el operador hamiltoniano para un procesamiento más eficiente en computadoras cuánticas, mejorando la eficiencia de la simulación. La estimación de la fase cuántica, por otro lado, ayuda a determinar con precisión los estados propios de energía, que son fundamentales para comprender el comportamiento del sistema cuántico. [77]
Si bien estas técnicas han hecho avanzar el campo de la química computacional, especialmente en la simulación de sistemas químicos, su aplicación práctica actualmente se limita principalmente a sistemas más pequeños debido a limitaciones tecnológicas. Sin embargo, estos avances pueden conducir a avances significativos hacia la consecución de simulaciones de química cuántica más precisas y eficientes en el uso de recursos. [76]
El costo computacional y la complejidad algorítmica en química se utilizan para ayudar a comprender y predecir fenómenos químicos. Ayudan a determinar qué algoritmos/métodos computacionales utilizar al resolver problemas químicos. Esta sección se centra en la escala de la complejidad computacional con el tamaño de la molécula y detalla los algoritmos comúnmente utilizados en ambos dominios. [78]
En química cuántica, particularmente, la complejidad puede crecer exponencialmente con el número de electrones involucrados en el sistema. Este crecimiento exponencial es una barrera importante para simular con precisión sistemas grandes o complejos. [79]
Los algoritmos avanzados en ambos campos se esfuerzan por equilibrar la precisión con la eficiencia computacional. Por ejemplo, en MD, se emplean métodos como la integración de Verlet o el algoritmo de Beeman por su eficiencia computacional. En química cuántica, los métodos híbridos que combinan diferentes enfoques computacionales (como QM/MM) se utilizan cada vez más para abordar grandes sistemas biomoleculares. [80]
La siguiente lista ilustra el impacto de la complejidad computacional en los algoritmos utilizados en cálculos químicos. Es importante señalar que, si bien esta lista proporciona ejemplos clave, no es exhaustiva y sirve como guía para comprender cómo las demandas computacionales influyen en la selección de métodos computacionales específicos en química.
Resuelve las ecuaciones de movimiento de Newton para átomos y moléculas. [81]
El cálculo estándar de interacción por pares en MD genera complejidad para las partículas. Esto se debe a que cada partícula interactúa con todas las demás, lo que resulta en interacciones. [82] Los algoritmos avanzados, como la suma de Ewald o el método rápido multipolar, reducen esto o incluso agrupan partículas distantes y las tratan como una sola entidad o utilizan aproximaciones matemáticas inteligentes. [83] [84]
Combina cálculos de mecánica cuántica para una región pequeña con mecánica molecular para un entorno más amplio. [85]
La complejidad de los métodos QM/MM depende tanto del tamaño de la región cuántica como del método utilizado para los cálculos cuánticos. Por ejemplo, si se utiliza un método de Hartree-Fock para la parte cuántica, la complejidad se puede aproximar como , donde es el número de funciones básicas en la región cuántica. Esta complejidad surge de la necesidad de resolver un conjunto de ecuaciones acopladas de forma iterativa hasta lograr la autoconsistencia. [86]

Encuentra un único estado de Fock que minimiza la energía. [87]
NP-duro o NP-completo como lo demuestra la incorporación de instancias del modelo de Ising en los cálculos de Hartree-Fock. El método Hartree-Fock implica resolver las ecuaciones de Roothaan-Hall, que escalan según la implementación, siendo el número de funciones básicas. El costo computacional proviene principalmente de evaluar y transformar las integrales de dos electrones. Esta prueba de dureza NP o completitud NP proviene de incorporar problemas como el modelo de Ising en el formalismo de Hartree-Fock. [87]
Investiga la estructura electrónica o la estructura nuclear de sistemas de muchos cuerpos, como átomos, moléculas y fases condensadas . [89]
Las implementaciones tradicionales de DFT suelen escalar como , principalmente debido a la necesidad de diagonalizar la matriz de Kohn-Sham . [90] El paso de diagonalización, que encuentra los valores propios y los vectores propios de la matriz, es el que más contribuye a este escalado. Los avances recientes en DFT tienen como objetivo reducir esta complejidad mediante diversas aproximaciones y mejoras algorítmicas. [91]
Los métodos CCSD y CCSD(T) son técnicas avanzadas de estructura electrónica que implican excitaciones simples, dobles y, en el caso de CCSD(T), triples perturbativas para calcular los efectos de correlación electrónica. [92]
Se escala como donde está el número de funciones básicas. Esta intensa demanda computacional surge de la inclusión de excitaciones simples y dobles en el cálculo de la correlación electrónica. [92]
Con la adición de tripletas perturbativas, la complejidad aumenta a . Esta elevada complejidad restringe el uso práctico a sistemas más pequeños, normalmente de hasta 20 a 25 átomos en implementaciones convencionales. [92]

Una adaptación del método CCSD(T) estándar que utiliza orbitales naturales (NO) locales para reducir significativamente la carga computacional y permitir la aplicación a sistemas más grandes. [92]
Logra un escalamiento lineal con el tamaño del sistema, una mejora importante con respecto al escalado tradicional de quinta potencia de CCSD. Este avance permite aplicaciones prácticas a moléculas de hasta 100 átomos con conjuntos de bases razonables, lo que marca un importante paso adelante en la capacidad de la química computacional para manejar sistemas más grandes con alta precisión. [92]
Demostrar las clases de complejidad de los algoritmos implica una combinación de prueba matemática y experimentos computacionales. Por ejemplo, en el caso del método Hartree-Fock, la prueba de la dureza NP es un resultado teórico derivado de la teoría de la complejidad, específicamente a través de reducciones de problemas NP-difíciles conocidos. [93]
Para otros métodos como MD o DFT, la complejidad computacional a menudo se observa empíricamente y se respalda mediante análisis de algoritmos. En estos casos, la prueba de corrección tiene menos que ver con pruebas matemáticas formales y más con observar consistentemente el comportamiento computacional en varios sistemas e implementaciones. [93]
La química computacional no es una descripción exacta de la química de la vida real, ya que los modelos matemáticos y físicos de la naturaleza sólo pueden proporcionar una aproximación. Sin embargo, la mayoría de los fenómenos químicos pueden describirse hasta cierto punto en un esquema computacional cualitativo o cuantitativo aproximado. [94]
Las moléculas están formadas por núcleos y electrones, por lo que se aplican los métodos de la mecánica cuántica . Los químicos computacionales a menudo intentan resolver la ecuación de Schrödinger no relativista , con correcciones relativistas añadidas, aunque se han logrado algunos avances en la resolución de la ecuación de Dirac totalmente relativista . En principio, es posible resolver la ecuación de Schrödinger en su forma dependiente o independiente del tiempo, según corresponda al problema en cuestión; en la práctica, esto no es posible excepto en sistemas muy pequeños. Por lo tanto, una gran cantidad de métodos aproximados se esfuerzan por lograr el mejor equilibrio entre precisión y costo computacional. [95]
La precisión siempre se puede mejorar con un mayor coste computacional. Pueden presentarse errores significativos en modelos ab initio que comprenden muchos electrones, debido al costo computacional de los métodos totalmente relativistas e inclusivos. [92] Esto complica el estudio de las moléculas que interactúan con átomos unitarios de alta masa atómica, como los metales de transición y sus propiedades catalíticas. Los algoritmos actuales en química computacional pueden calcular de forma rutinaria las propiedades de moléculas pequeñas que contienen hasta aproximadamente 40 electrones con errores para energías inferiores a unos pocos kJ/mol. Para las geometrías, las longitudes de los enlaces se pueden predecir dentro de unos pocos picómetros y los ángulos de los enlaces dentro de 0,5 grados. El tratamiento de moléculas más grandes que contienen unas pocas docenas de átomos es computacionalmente manejable mediante métodos más aproximados, como la teoría funcional de la densidad (DFT). [96]
Existe cierta controversia dentro del campo sobre si estos últimos métodos son suficientes o no para describir reacciones químicas complejas, como las de la bioquímica. Las moléculas grandes se pueden estudiar mediante métodos aproximados semiempíricos. Incluso las moléculas más grandes se tratan mediante métodos de mecánica clásica que utilizan lo que se llama mecánica molecular (MM). En los métodos QM-MM, las partes pequeñas de grandes complejos se tratan de forma cuántica (QM) y el resto se trata de forma aproximada (MM). [97]
Existen muchos paquetes de software de química computacional autosuficientes . Algunos incluyen muchos métodos que cubren una amplia gama, mientras que otros se concentran en un rango muy específico o incluso en un método. Los detalles de la mayoría de ellos se pueden encontrar en:











