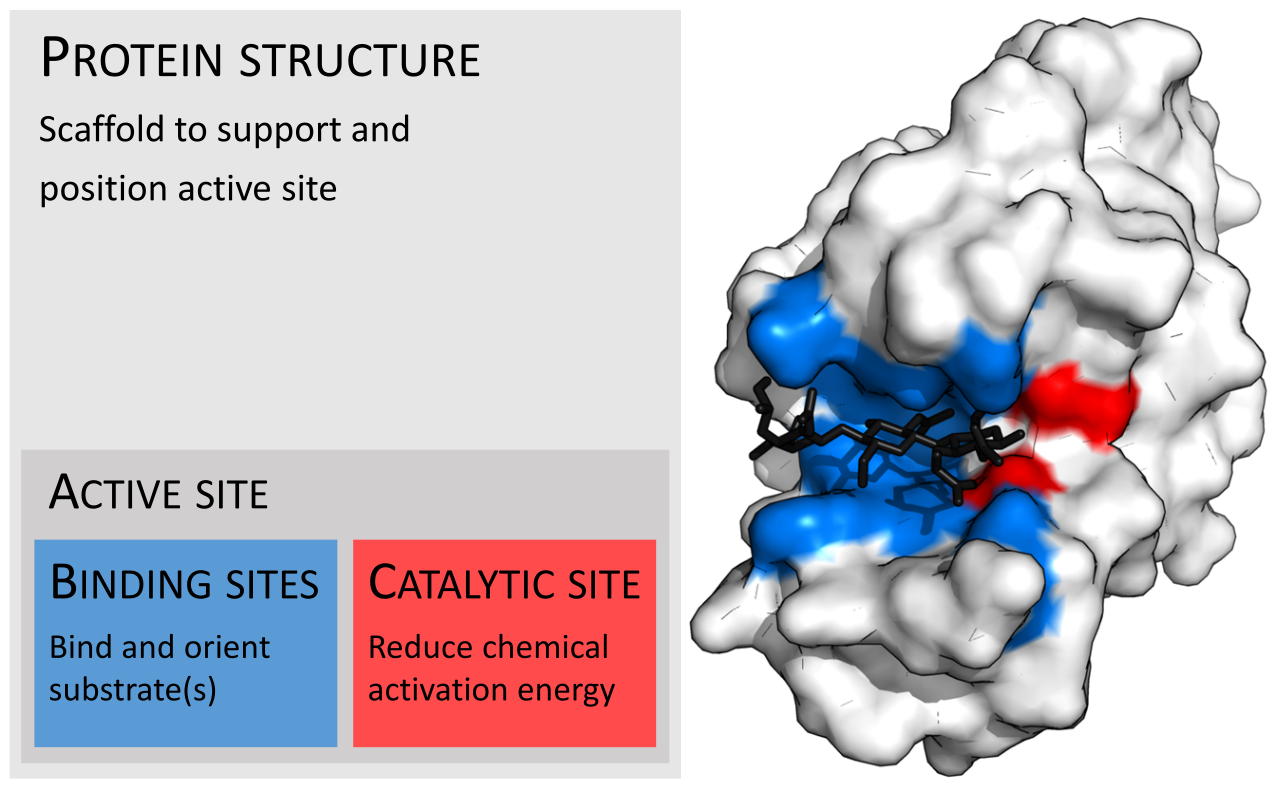
En biología y bioquímica , el sitio activo es la región de una enzima donde las moléculas de sustrato se unen y experimentan una reacción química. El sitio activo consta de residuos de aminoácidos que forman enlaces temporales con el sustrato, el sitio de unión , y residuos que catalizan una reacción de ese sustrato, el sitio catalítico . Aunque el sitio activo ocupa solo ~10–20% del volumen de una enzima, [1] : 19 es la parte más importante ya que cataliza directamente la reacción química . Por lo general, consta de tres a cuatro aminoácidos, mientras que otros aminoácidos dentro de la proteína son necesarios para mantener la estructura terciaria de las enzimas. [2]
Cada sitio activo evoluciona para optimizarse para unirse a un sustrato particular y catalizar una reacción particular, lo que resulta en una alta especificidad . Esta especificidad está determinada por la disposición de los aminoácidos dentro del sitio activo y la estructura de los sustratos. A veces, las enzimas también necesitan unirse con algunos cofactores para cumplir su función. El sitio activo suele ser un surco o bolsillo de la enzima que puede estar ubicado en un túnel profundo dentro de la enzima, [3] o entre las interfaces de enzimas multiméricas . Un sitio activo puede catalizar una reacción repetidamente ya que los residuos no se alteran al final de la reacción (pueden cambiar durante la reacción, pero se regeneran al final). [4] Este proceso se logra reduciendo la energía de activación de la reacción, por lo que más sustratos tienen suficiente energía para experimentar la reacción.
Por lo general, una molécula de enzima tiene un solo sitio activo, y este sitio activo se adapta a un tipo específico de sustrato. Un sitio activo contiene un sitio de unión que une el sustrato y lo orienta para la catálisis. La orientación del sustrato y la proximidad entre este y el sitio activo son tan importantes que en algunos casos la enzima puede seguir funcionando correctamente aunque todas las demás partes hayan mutado y pierdan su función. [5]
Inicialmente, la interacción entre el sitio activo y el sustrato no es covalente y transitoria. Hay cuatro tipos importantes de interacción que mantienen al sustrato en una orientación definida y forman un complejo enzima-sustrato (complejo ES): enlaces de hidrógeno , interacciones de van der Waals , interacciones hidrofóbicas e interacciones de fuerza electrostática . [6] : 148 La distribución de carga en el sustrato y el sitio activo debe ser complementaria, lo que significa que todas las cargas positivas y negativas deben cancelarse. De lo contrario, habrá una fuerza repulsiva que los separe. El sitio activo generalmente contiene aminoácidos no polares , aunque a veces también pueden aparecer aminoácidos polares. [2] La unión del sustrato al sitio de unión requiere al menos tres puntos de contacto para lograr estereo-, regio- y enantioselectividad. Por ejemplo, la alcohol deshidrogenasa que cataliza la transferencia de un ion hidruro de etanol a NAD + interactúa con el grupo metilo del sustrato , el grupo hidroxilo y el hidrógeno pro- (R) que se abstraerá durante la reacción. [6] : 149
Para ejercer su función, las enzimas necesitan asumir su plegamiento proteico correcto ( plegamiento nativo ) y su estructura terciaria . Para mantener esta estructura tridimensional definida, las proteínas dependen de varios tipos de interacciones entre sus residuos de aminoácidos. Si estas interacciones se ven interferidas, por ejemplo, por valores de pH extremos, altas temperaturas o altas concentraciones de iones, esto hará que la enzima se desnaturalice y pierda su actividad catalítica. [ cita requerida ]
Se cree que un ajuste más estrecho entre un sitio activo y la molécula de sustrato aumenta la eficiencia de una reacción. Si aumenta la estrechez entre el sitio activo de la ADN polimerasa y su sustrato, también aumentará la fidelidad, es decir, la tasa correcta de replicación del ADN. [7] La mayoría de las enzimas tienen sitios activos enterrados profundamente, a los que un sustrato puede acceder a través de canales de acceso. [3]
Existen tres modelos propuestos de cómo las enzimas se adaptan a su sustrato específico: el modelo de llave y cerradura , el modelo de ajuste inducido y el modelo de selección conformacional. Los dos últimos no son mutuamente excluyentes: la selección conformacional puede ser seguida por un cambio en la forma de la enzima. Además, una proteína puede no seguir completamente ninguno de los dos modelos. Los aminoácidos en el sitio de unión de la ubiquitina generalmente siguen el modelo de ajuste inducido, mientras que el resto de la proteína generalmente se adhiere a la selección conformacional. Es probable que factores como la temperatura influyan en la vía tomada durante la unión, y se predice que las temperaturas más altas aumentarán la importancia de la selección conformacional y disminuirán la del ajuste inducido. [8]
Este concepto fue sugerido por el químico del siglo XIX Emil Fischer . Propuso que el sitio activo y el sustrato son dos estructuras estables que encajan perfectamente sin ninguna modificación adicional, tal como una llave encaja en una cerradura. Si un sustrato se une perfectamente a su sitio activo, las interacciones entre ellos serán más fuertes, lo que dará como resultado una alta eficiencia catalítica.
Con el paso del tiempo, este modelo empezó a tener sus limitaciones. Por ejemplo, el inhibidor enzimático competitivo metilglucósido puede unirse fuertemente al sitio activo de la 4-alfa-glucanotransferasa y encaja perfectamente en él. Sin embargo, la 4-alfa-glucanotransferasa no es activa sobre el metilglucósido y no se produce transferencia de glicosilo. La hipótesis de la cerradura y la llave no puede explicar esto, ya que predeciría una alta eficiencia de la transferencia de glicosilo del metilglucósido debido a su fuerte unión. Aparte de la inhibición competitiva, esta teoría tampoco puede explicar el mecanismo de acción de los inhibidores no competitivos , ya que no se unen al sitio activo pero, sin embargo, influyen en la actividad catalítica. [9]
La teoría de Daniel Koshland sobre la unión enzima-sustrato es que el sitio activo y la porción de unión del sustrato no son exactamente complementarios. [10] El modelo de ajuste inducido es un desarrollo del modelo de llave y cerradura y supone que un sitio activo es flexible y cambia de forma hasta que el sustrato está completamente unido. Este modelo es similar a una persona que usa un guante: el guante cambia de forma para adaptarse a la mano. La enzima inicialmente tiene una conformación que atrae a su sustrato. La superficie de la enzima es flexible y solo el catalizador correcto puede inducir la interacción que conduce a la catálisis. Luego pueden ocurrir cambios conformacionales a medida que se une el sustrato. Después, los productos de la reacción se alejarán de la enzima y el sitio activo volverá a su forma inicial. Esta hipótesis está respaldada por la observación de que todo el dominio proteico podría moverse varios nanómetros durante la catálisis. Este movimiento de la superficie de la proteína puede crear microambientes que favorezcan la catálisis. [5]
Este modelo sugiere que las enzimas existen en una variedad de conformaciones, de las cuales solo algunas son capaces de unirse a un sustrato. Cuando un sustrato se une a la proteína, el equilibrio en el conjunto conformacional se desplaza hacia aquellas capaces de unirse a ligandos (ya que las enzimas con sustratos unidos se eliminan del equilibrio entre las conformaciones libres). [11]
Interacción electrostática : en un entorno acuoso, los grupos con carga opuesta en las cadenas laterales de aminoácidos dentro del sitio activo y los sustratos se atraen entre sí, lo que se denomina interacción electrostática. Por ejemplo, cuando un ácido carboxílico (R-COOH) se disocia en iones RCOO − y H + , COO − atraerá grupos con carga positiva, como la cadena lateral de guanidina protonada de la arginina . [ cita requerida ]
Enlace de hidrógeno : un enlace de hidrógeno es un tipo específico de interacción dipolo-dipolo entre un átomo de hidrógeno parcialmente positivo y un donador de electrones parcialmente negativo que contiene un par de electrones como el oxígeno , el flúor y el nitrógeno . La fuerza del enlace de hidrógeno depende de la naturaleza química y la disposición geométrica de cada grupo. [ cita requerida ]
Fuerza de Van der Waals : la fuerza de Van der Waals se forma entre grupos con cargas opuestas debido a la distribución desigual transitoria de los electrones en cada grupo. Si todos los electrones están concentrados en un polo del grupo, este extremo será negativo, mientras que el otro extremo será positivo. Aunque la fuerza individual es débil, como el número total de interacciones entre el sitio activo y el sustrato es enorme, la suma de ellas será significativa. [ cita requerida ]
Interacción hidrofóbica : los grupos hidrofóbicos no polares tienden a agruparse en un entorno acuoso y tratan de salir del disolvente polar. Estos grupos hidrofóbicos suelen tener una cadena de carbono larga y no reaccionan con las moléculas de agua. Al disolverse en agua, una molécula de proteína se enroscará en una forma similar a una bola, dejando los grupos hidrofílicos en el exterior, mientras que los grupos hidrofóbicos quedan enterrados profundamente en el centro. [ cita requerida ]

Una vez que el sustrato está unido y orientado al sitio activo, puede comenzar la catálisis . Los residuos del sitio catalítico suelen estar muy cerca del sitio de unión y algunos residuos pueden tener una doble función, tanto en la unión como en la catálisis. [ cita requerida ]
Los residuos catalíticos del sitio interactúan con el sustrato para reducir la energía de activación de una reacción y, por lo tanto, hacer que avance más rápido . Lo hacen mediante una serie de mecanismos diferentes, que incluyen la aproximación de los reactivos, la catálisis nucleofílica/electrófila y la catálisis ácido/base. Estos mecanismos se explicarán a continuación. [ cita requerida ]
Durante la reacción catalítica enzimática, el sustrato y el sitio activo se juntan en una proximidad cercana. Este enfoque tiene varios propósitos. En primer lugar, cuando los sustratos se unen dentro del sitio activo, la concentración efectiva de este aumenta significativamente que en solución. Esto significa que el número de moléculas de sustrato involucradas en la reacción también aumenta. Este proceso también reduce la energía de desolvatación requerida para que se produzca la reacción. En solución, las moléculas de sustrato están rodeadas por moléculas de disolvente y se requiere energía para que las moléculas de enzima las reemplacen y entren en contacto con el sustrato. Dado que las moléculas a granel se pueden excluir del sitio activo, esta salida de energía se puede minimizar. A continuación, el sitio activo está diseñado para reorientar el sustrato para reducir la energía de activación para que se produzca la reacción. La alineación del sustrato, después de la unión, se bloquea en un estado de alta energía y puede continuar al siguiente paso. Además, esta unión se ve favorecida por la entropía , ya que el costo de energía asociado con la reacción en solución se elimina en gran medida, ya que el disolvente no puede ingresar al sitio activo. Al final, el sitio activo puede manipular el orbital molecular del sustrato en una orientación adecuada para reducir la energía de activación. [6] : 155–8
Los estados electrostáticos del sustrato y del sitio activo deben ser complementarios entre sí. Una cadena lateral de aminoácidos polarizada y cargada negativamente repelerá un sustrato no cargado. Pero si el estado de transición implica la formación de un centro iónico , entonces la cadena lateral producirá una interacción favorable.
Muchas enzimas, incluidas la serina proteasa , la cisteína proteasa , la proteína quinasa y la fosfatasa, evolucionaron para formar enlaces covalentes transitorios entre ellas y sus sustratos para reducir la energía de activación y permitir que se produzca la reacción. Este proceso se puede dividir en dos pasos: formación y descomposición. El primer paso es el paso límite de velocidad, mientras que el último paso es necesario para regenerar la enzima intacta. [6] : 158
Catálisis nucleofílica : Este proceso implica la donación de electrones del nucleófilo de la enzima a un sustrato para formar un enlace covalente entre ellos durante el estado de transición. La fuerza de esta interacción depende de dos aspectos: la capacidad del grupo nucleófilo para donar electrones y del electrófilo para aceptarlos. El primero se ve afectado principalmente por la basicidad (la capacidad de donar pares de electrones) de la especie, mientras que el segundo se ve afectado por su p K a . Ambos grupos también se ven afectados por sus propiedades químicas, como la polarizabilidad , la electronegatividad y el potencial de ionización . Los aminoácidos que pueden formar nucleófilos incluyen serina , cisteína , aspartato y glutamina . [ cita requerida ]
Catálisis electrofílica : el mecanismo detrás de este proceso es exactamente el mismo que el de la catálisis nucleofílica, excepto que ahora los aminoácidos en el sitio activo actúan como electrófilos mientras que los sustratos son nucleófilos . Esta reacción generalmente requiere cofactores ya que las cadenas laterales de los aminoácidos no son lo suficientemente fuertes para atraer electrones.
Los iones metálicos tienen múltiples funciones durante la reacción. En primer lugar, pueden unirse a grupos de sustrato con carga negativa para que no repelan pares de electrones de los grupos nucleofílicos del sitio activo. Pueden atraer electrones con carga negativa para aumentar la electrofilia . También pueden hacer de puente entre el sitio activo y el sustrato. Por último, pueden cambiar la estructura conformacional del sustrato para favorecer la reacción. [6] : 158
En algunas reacciones, los protones y el hidróxido pueden actuar directamente como ácido y base en términos de catálisis de ácido específico y base específica. Pero más a menudo los grupos en el sustrato y el sitio activo actúan como ácido y base de Brønsted-Lowry. Esto se llama teoría general de ácido y base general. La forma más fácil de distinguirlos es verificar si la velocidad de reacción está determinada por las concentraciones del ácido y base generales. Si la respuesta es sí, entonces la reacción es del tipo general. Dado que la mayoría de las enzimas tienen un pH óptimo de 6 a 7, los aminoácidos en la cadena lateral generalmente tienen un p K a de 4 ~ 10. Los candidatos incluyen aspartato , glutamato , histidina , cisteína . Estos ácidos y bases pueden estabilizar el nucleófilo o electrófilo formado durante la catálisis al proporcionar cargas positivas y negativas. [6] : 164–70
Los estudios cuantitativos de las reacciones enzimáticas han demostrado a menudo que la aceleración de la velocidad de las reacciones químicas no puede explicarse por completo con las teorías existentes, como la aproximación, la catálisis ácido/base y la catálisis electrófila/nucleófila. Y existe una paradoja obvia: en una reacción enzimática reversible, si el sitio activo encaja perfectamente con los sustratos, la reacción inversa se ralentizará, ya que los productos no pueden encajar perfectamente en el sitio activo. Por eso se introdujo la distorsión conformacional, que sostiene que tanto el sitio activo como el sustrato pueden sufrir cambios conformacionales para encajar entre sí todo el tiempo. [6] : 170–5
Esta teoría es un poco similar a la teoría de la cerradura y la llave, pero en este caso el sitio activo está preprogramado para unirse perfectamente al sustrato en estado de transición en lugar de en estado fundamental. La formación del estado de transición dentro de la solución requiere una gran cantidad de energía para reubicar las moléculas de disolvente y la reacción se ralentiza. Por lo tanto, el sitio activo puede sustituir a las moléculas de disolvente y rodear a los sustratos para minimizar el efecto contraproducente impuesto por la solución. La presencia de grupos cargados con el sitio activo atraerá a los sustratos y garantizará la complementariedad electrostática. [6] : 176–8
En realidad, la mayoría de los mecanismos enzimáticos implican una combinación de varios tipos diferentes de catálisis.
La función del glutatión (GSH) es eliminar las especies reactivas de oxígeno acumuladas que pueden dañar las células. Durante este proceso, su cadena lateral tiol se oxida y dos moléculas de glutatión se conectan mediante un enlace disulfuro para formar un dímero (GSSG). Para regenerar el glutatión, el enlace disulfuro debe romperse. En las células humanas, esto lo hace la glutatión reductasa (GR). [ cita requerida ]
La glutatión reductasa es un dímero que contiene dos subunidades idénticas. Requiere un NADP y un FAD como cofactores . El sitio activo se encuentra en el enlace entre dos subunidades. El NADPH está involucrado en la generación de FADH-. En el sitio activo, hay dos residuos de cisteína además del cofactor FAD y se utilizan para romper el enlace disulfuro durante la reacción catalítica. El NADPH está unido por tres residuos con carga positiva: Arg-218, His-219 y Arg-224. [ cita requerida ]
El proceso catalítico comienza cuando el FAD es reducido por NADPH para aceptar un electrón y del FADH − . Luego ataca el enlace disulfuro formado entre 2 residuos de cisteína, formando un enlace SH y un solo grupo S − . Este grupo S − actuará como un nucleófilo para atacar el enlace disulfuro en el glutatión oxidado (GSSG), rompiéndolo y formando un complejo cisteína-SG. El primer anión SG − se libera y luego recibe un protón del grupo SH adyacente y del primer monómero de glutatión. A continuación, el grupo S − adyacente ataca el enlace disulfuro en el complejo cisteína-SG y libera el segundo anión SG − . Recibe un protón en solución y forma el segundo monómero de glutatión.
[1] : 137–9

La quimotripsina es una endopeptidasa de serina que está presente en el jugo pancreático y ayuda a la hidrólisis de proteínas y péptidos . [1] : 84–6 Cataliza la hidrólisis de enlaces peptídicos en los isómeros L de tirosina , fenilalanina y triptófano . En el sitio activo de esta enzima, tres residuos de aminoácidos trabajan juntos para formar una tríada catalítica que constituye el sitio catalítico. En la quimotripsina, estos residuos son Ser-195, His-57 y Asp-102.
El mecanismo de la quimotripsina se puede dividir en dos fases. En primer lugar, Ser-195 ataca nucleofílicamente el carbono del enlace peptídico en el sustrato para formar un intermediario tetraédrico. La nucleofilia de Ser-195 se ve potenciada por His-57, que abstrae un protón de Ser-195 y, a su vez, se estabiliza mediante el grupo carboxilato con carga negativa (RCOO − ) en Asp-102. Además, el intermediario oxianión tetraédrico generado en este paso se estabiliza mediante enlaces de hidrógeno de Ser-195 y Gly-193.
En la segunda etapa, el grupo R'NH es protonado por His-57 para formar R'NH2 y abandona el intermediario, dejando atrás la Ser-195 acilada . His-57 actúa entonces como una base nuevamente para abstraer un protón de una molécula de agua. El anión hidróxido resultante ataca nucleofílicamente al complejo acil-enzima para formar un segundo intermediario oxianión tetraédrico, que es nuevamente estabilizado por enlaces de H. Al final, Ser-195 abandona el intermediario tetraédrico, rompiendo el enlace CO que conectaba la enzima al sustrato peptídico. Un protón es transferido a Ser-195 a través de His-57, de modo que los tres aminoácidos regresan a su estado inicial.
La desunión del sustrato está influenciada por varios factores. Los ligandos más grandes generalmente permanecen en el sitio activo por más tiempo, [12] al igual que aquellos con enlaces más rotativos (aunque esto puede ser un efecto secundario del tamaño). [13] Cuando el solvente se excluye del sitio activo, las proteínas menos flexibles dan como resultado tiempos de residencia más largos . Más enlaces de hidrógeno protegidos del solvente también disminuyen la desunión. [12]

Las enzimas pueden usar cofactores como 'moléculas auxiliares'. Las coenzimas son aquellas moléculas no proteicas que se unen a las enzimas para ayudarlas a cumplir con su trabajo. En su mayoría están conectadas al sitio activo por enlaces no covalentes como el enlace de hidrógeno o la interacción hidrofóbica . Pero a veces también se puede formar un enlace covalente entre ellas. Por ejemplo, el hemo en el citocromo C está unido a la proteína a través del enlace tioéster . En algunas ocasiones, las coenzimas pueden abandonar las enzimas una vez finalizada la reacción. De lo contrario, se unen permanentemente a la enzima. [6] : 69 La coenzima es un concepto amplio que incluye iones metálicos, varias vitaminas y ATP . Si una enzima necesita coenzima para funcionar por sí misma, se denomina apoenzima. De hecho, por sí sola no puede catalizar las reacciones adecuadamente. Solo cuando su cofactor entra y se une al sitio activo para formar la holoenzima, funciona correctamente.
Un ejemplo de coenzima es la flavina . Contiene un sistema de anillo de isoaloxazina conjugada distintivo. La flavina tiene múltiples estados redox y se puede utilizar en procesos que implican la transferencia de uno o dos electrones. Puede actuar como aceptor de electrones en una reacción, como la oxidación de NAD a NADH, para aceptar dos electrones y formar 1,5-dihidroflavina. Por otro lado, puede formar semiquinona ( radical libre ) al aceptar un electrón, y luego convertirse en una forma completamente reducida mediante la adición de un electrón adicional. Esta propiedad le permite ser utilizada en un proceso de oxidación de un electrón.
Los inhibidores alteran la interacción entre la enzima y el sustrato, lo que ralentiza la velocidad de una reacción. Existen distintos tipos de inhibidores, tanto reversibles como irreversibles.
Los inhibidores competitivos son inhibidores que solo se dirigen a las moléculas de enzimas libres. Compiten con los sustratos por el aceptor de enzimas libres y se pueden superar aumentando la concentración de sustrato. Tienen dos mecanismos. Los inhibidores competitivos suelen tener similitudes estructurales con los sustratos o el complejo ES. Como resultado, pueden encajar en el sitio activo y desencadenar interacciones favorables para rellenar el espacio y bloquear la entrada de sustratos. También pueden inducir cambios conformacionales transitorios en el sitio activo para que los sustratos no puedan encajar perfectamente en él. Después de un corto período de tiempo, los inhibidores competitivos se desprenderán y dejarán la enzima intacta.
Los inhibidores se clasifican como inhibidores no competitivos cuando se unen a la enzima libre y al complejo ES. Dado que no compiten con los sustratos por el sitio activo, no se los puede superar simplemente aumentando la concentración del sustrato. Por lo general, se unen a un sitio diferente de la enzima y alteran la estructura tridimensional del sitio activo para bloquear la entrada o salida de los sustratos de la enzima.
Los inhibidores irreversibles son similares a los inhibidores competitivos, ya que ambos se unen al sitio activo. Sin embargo, los inhibidores irreversibles forman enlaces covalentes irreversibles con los residuos de aminoácidos en el sitio activo y nunca se van. Por lo tanto, el sitio activo está ocupado y el sustrato no puede entrar. Ocasionalmente, el inhibidor se irá, pero el sitio catalítico se altera permanentemente en su forma. Estos inhibidores suelen contener grupos electrófilos como sustitutos de halógenos y epóxidos . A medida que pasa el tiempo, cada vez más enzimas se unen a inhibidores irreversibles y ya no pueden funcionar.

Los inhibidores de la proteasa del VIH se utilizan para tratar a los pacientes con el virus del SIDA , impidiendo la replicación de su ADN . El virus utiliza la proteasa del VIH para escindir la poliproteína Gag-Pol en tres proteínas más pequeñas que son responsables del ensamblaje, empaquetamiento y maduración del virión. Esta enzima se dirige al sitio de escisión específico de fenilalanina - prolina dentro de la proteína diana. [14] Si se desactiva la proteasa del VIH, la partícula del virión perderá su función y no podrá infectar a los pacientes. Dado que es esencial en la replicación viral y está ausente en humanos sanos, es un objetivo ideal para el desarrollo de fármacos .
La proteasa del VIH pertenece a la familia de las proteasas aspárticas y tiene un mecanismo similar. En primer lugar, el residuo de aspartato activa una molécula de agua y la convierte en un nucleófilo . Luego ataca al grupo carbonilo dentro del enlace peptídico (NH-CO) para formar un intermediario tetraédrico. El átomo de nitrógeno dentro del intermediario recibe un protón, formando un grupo amida y la posterior reorganización conduce a la ruptura del enlace entre él y el intermediario y forma dos productos. [15]
Los inhibidores suelen contener grupos hidroxietileno o hidroxietilamina no hidrolizables que imitan al intermediario tetraédrico. Dado que comparten una estructura y una disposición electrostática similares al estado de transición de los sustratos, pueden encajar en el sitio activo pero no pueden descomponerse, por lo que no puede producirse la hidrólisis.
La estricnina es una neurotoxina que causa la muerte al afectar los nervios que controlan la contracción muscular y causar dificultad respiratoria. El impulso se transmite entre las sinapsis a través de un neurotransmisor llamado acetilcolina . Se libera en la sinapsis entre las células nerviosas y se une a los receptores de la célula postsináptica. Luego se genera un potencial de acción y se transmite a través de la célula postsináptica para iniciar un nuevo ciclo.
La glicina puede inhibir la actividad de los receptores de neurotransmisores, por lo que se requiere una mayor cantidad de acetilcolinesterasa para desencadenar un potencial de acción. Esto garantiza que la generación de impulsos nerviosos esté estrictamente controlada. Sin embargo, este control se rompe cuando se agrega estricnina. Inhibe los receptores de glicina (un canal de cloruro ) y un nivel mucho menor de concentración de neurotransmisores puede desencadenar un potencial de acción. Los nervios ahora transmiten señales constantemente y causan una contracción muscular excesiva, lo que lleva a la asfixia y la muerte. [16]

El diisopropil fluorofosfato (DIFP) es un inhibidor irreversible que bloquea la acción de la serina proteasa . Cuando se une a la enzima, se produce una reacción de sustitución nucleofílica y se libera una molécula de fluoruro de hidrógeno . El grupo OH en el sitio activo actúa como un nucleófilo para atacar el fósforo en DIFP y formar un intermediario tetraédrico y liberar un protón. Luego, el enlace PF se rompe, un electrón se transfiere al átomo de F y abandona el intermediario como anión F − . Se combina con un protón en solución para formar una molécula de HF. Se forma un enlace covalente entre el sitio activo y DIFP, por lo que la cadena lateral de serina ya no está disponible para el sustrato. [17]
La identificación de los sitios activos es crucial en el proceso de descubrimiento de fármacos . La estructura tridimensional de la enzima se analiza para identificar los residuos del sitio activo y diseñar fármacos que puedan encajar en ellos. Las enzimas proteolíticas son objetivos de algunos fármacos, como los inhibidores de la proteasa, que incluyen fármacos contra el SIDA y la hipertensión. [18] Estos inhibidores de la proteasa se unen al sitio activo de una enzima y bloquean la interacción con sustratos naturales. [19] Un factor importante en el diseño de fármacos es la fuerza de unión entre el sitio activo y un inhibidor de enzima. [20] Si la enzima que se encuentra en las bacterias es significativamente diferente de la enzima humana, entonces se puede diseñar un inhibidor contra esa bacteria en particular sin dañar la enzima humana. Si un tipo de enzima solo está presente en un tipo de organismo, su inhibidor se puede utilizar para eliminarlos específicamente.
Los sitios activos se pueden mapear para ayudar al diseño de nuevos fármacos, como los inhibidores de enzimas. Esto implica la descripción del tamaño de un sitio activo y la cantidad y las propiedades de los subsitios, como los detalles de la interacción de unión. [18] Sin embargo, la tecnología de base de datos moderna llamada CPASS (Comparación de estructuras de sitios activos de proteínas) permite la comparación de sitios activos con más detalle y el hallazgo de similitudes estructurales mediante software. [21]

Un sitio alostérico es un sitio en una enzima, no relacionado con su sitio activo, que puede unirse a una molécula efectora. Esta interacción es otro mecanismo de regulación enzimática. La modificación alostérica suele ocurrir en proteínas con más de una subunidad. Las interacciones alostéricas suelen estar presentes en las vías metabólicas y son beneficiosas porque permiten que un paso de una reacción regule otro paso. [19] Permiten que una enzima tenga una variedad de interacciones moleculares, además del sitio activo altamente específico. [19]
{kind=link}