La osteogénesis imperfecta ( IPA : / ˌ ɒ s t i oʊ ˈ dʒ ɛ n ə s ɪ s ˌ ɪ m p ɜːr ˈ f ɛ k t ə / ; [4] OI ), conocida coloquialmente como enfermedad de los huesos de cristal , es un grupo de Trastornos genéticos que resultan en huesos que se rompen fácilmente. [1] : 85 [9] La variedad de síntomas, tanto en el esqueleto como en otros órganos del cuerpo , puede ser de leve a grave. [5] : 1512 Los síntomas que se encuentran en varios tipos de OI incluyen la parte blanca de los ojos (escleróticas) que en cambio son azules, estatura baja , articulaciones flojas , pérdida de audición , problemas respiratorios [10] y problemas con los dientes ( dentinogénesis imperfecta ). [5] Las complicaciones potencialmente mortales , todas las cuales se vuelven más comunes en la OI más grave, incluyen: desgarro ( disección ) de las arterias principales , como la aorta ; [1] : 333 [11] insuficiencia de la válvula pulmonar secundaria a distorsión de la caja torácica ; [1] : 335–341 [12] e invaginación basilar . [13] : 106-107
El mecanismo subyacente suele ser un problema con el tejido conectivo debido a la falta de colágeno tipo I o su mala formación . [5] : 1513 En más del 90% de los casos, la OI se produce debido a mutaciones en los genes COL1A1 o COL1A2 . [14] Estas mutaciones pueden ser hereditarias de manera autosómica dominante , pero también pueden ocurrir espontáneamente ( de novo ). [9] [15] Hay cuatro tipos clínicamente definidos : tipo I, el menos grave; tipo IV, moderadamente grave; tipo III, grave y progresivamente deformante; y el tipo II, perinatalmente letal. [9] En septiembre de 2021 [actualizar], se sabe que 19 genes diferentes causan los 21 tipos de OI documentados genéticamente definidos, muchos de los cuales son extremadamente raros y solo se han documentado en unos pocos individuos. [16] [17] El diagnóstico a menudo se basa en los síntomas y puede confirmarse mediante una biopsia de colágeno o una secuenciación de ADN . [10]
Aunque no existe cura, [10] la mayoría de los casos de OI no tienen un efecto importante en la esperanza de vida, [1] : 461 [15] la muerte durante la niñez a causa de esta enfermedad es rara, [10] y muchos adultos con OI pueden lograr una Grado significativo de autonomía a pesar de la discapacidad . [18] Mantener un estilo de vida saludable haciendo ejercicio , llevando una dieta equilibrada con suficiente contenido de vitamina D y calcio y evitando fumar puede ayudar a prevenir fracturas. [19] Las personas con OI pueden solicitar asesoramiento genético para evitar que sus hijos hereden el trastorno. [1] : 101 El tratamiento puede incluir cuidados intensivos de huesos rotos, analgésicos , fisioterapia , ayudas para la movilidad como aparatos ortopédicos y sillas de ruedas , [10] suplementos de vitamina D y, especialmente en la infancia, cirugía con varillas . [20] Rodding es una implantación de varillas intramedulares de metal a lo largo de los huesos largos (como el fémur ) en un intento de fortalecerlos. [10] La investigación médica también respalda el uso de medicamentos de la clase de los bifosfonatos , como el pamidronato , para aumentar la densidad ósea . [21] Los bifosfonatos son especialmente eficaces en los niños, [22] sin embargo, no está claro si aumentan la calidad de vida o disminuyen la tasa de incidencia de fracturas. [7]
La OI afecta sólo a una de cada 15.000 a 20.000 personas, lo que la convierte en una enfermedad genética poco común . [8] Los resultados dependen de la causa genética del trastorno (su tipo). El tipo I (el menos grave) es el más común, y los otros tipos constituyen una minoría de los casos. [15] [23] [24] La OI de moderada a grave afecta principalmente la movilidad; Si la cirugía con varilla se realiza durante la niñez, algunas personas con tipos más graves de OI pueden adquirir la capacidad de caminar. [25] La condición ha sido descrita desde la historia antigua. [26] El término latino osteogénesis imperfecta fue acuñado por el anatomista holandés Willem Vrolik en 1849; Traducido literalmente, significa "formación ósea imperfecta". [26] [27] : 683

El principal síntoma de la osteogénesis imperfecta son los huesos frágiles y de baja densidad mineral ; Todos los tipos de OI tienen alguna afectación ósea. [5] En la OI moderada y especialmente grave, los huesos largos pueden estar arqueados , a veces en extremo. [28] La debilidad de los huesos hace que se fracturen fácilmente: un estudio realizado en la Unidad Endocrina del Instituto Nacional de Salud Infantil en Karachi, Pakistán, encontró un promedio de 5,8 fracturas por año en niños no tratados. [29] Las fracturas suelen ocurrir mucho menos después de la pubertad , pero comienzan a aumentar nuevamente en las mujeres después de la menopausia y en los hombres entre las edades de 60 y 80 años. [1] : 486
La hipermovilidad de las articulaciones también es un signo común de OI, probablemente porque los genes afectados son los mismos que causan algunos tipos de síndrome de Ehlers-Danlos . [5] : 1513 [nota 1] [30] [31]
A la edad de 50 años, aproximadamente el 50% de los adultos con OI experimentan una pérdida auditiva significativa , mucho antes en comparación con la población general. [32] La pérdida de audición en la OI puede estar asociada o no con deformidades visibles de los huesecillos y el oído interno . [33] La pérdida de audición frecuentemente comienza durante la segunda, tercera y cuarta décadas de la vida y puede ser conductiva , neurosensorial o una combinación de ambas ("mixta"). [34] Si la pérdida de audición no ocurre a los 50 años, es mucho menos probable que ocurra en los años posteriores. [32] La pérdida auditiva mixta es más común entre personas con OI de todos los grupos de edad, mientras que la pérdida auditiva conductiva es más probable que afecte a las personas mayores, y la pérdida auditiva neurosensorial es más probable que afecte a los niños. [35]
Aunque es relativamente rara, la pérdida auditiva relacionada con la OI también puede comenzar en la niñez; en un estudio de cuarenta y cinco niños de cuatro a dieciséis años, se encontró que dos estaban afectados, de 11 y 15 años. [36] En un estudio diferente de 2008, se revisó la audición de 41 personas con OI. Los resultados mostraron que el 88% de los mayores de 20 años presentaban algún tipo de pérdida auditiva, mientras que sólo el 38% de los menores de 20 años la padecían. [37]
La pérdida de audición es más común en la OI tipo I; es menos común en los tipos III y IV. [1] : 294–296 Otras partes del oído interno también pueden verse afectadas por la OI. causando problemas de equilibrio ; sin embargo, sólo estudios pequeños han encontrado vínculos entre el vértigo y la OI. [1] : 308 La IO puede empeorar el resultado de los tratamientos médicos que corrigen la pérdida auditiva. [35]
Además de la asociación de la OI con la pérdida auditiva neurosensorial, la OI se asocia con una serie de anomalías neurológicas, que generalmente afectan al sistema nervioso central , debido a deformidades en las estructuras esqueléticas que lo rodean. Las complicaciones neurológicas, especialmente la invaginación basilar , pueden afectar negativamente a la esperanza de vida. En la OI, esto se debe con mayor frecuencia a la migración hacia arriba de las madrigueras , [38] [13] : 106–107, una característica de la vértebra C2 . La neurocirugía puede ser necesaria para corregir anomalías graves cuando ponen en riesgo la vida del paciente o causan un gran sufrimiento o déficits neurológicos intolerables . [38] [13] : 106-107
A medida que sus causas biológicas se han determinado con mayor precisión, se ha reconocido más ampliamente que, si bien el proceso primario de la enfermedad de la OI ocurre en los huesos, los tipos más comunes de OI (aquellas causadas por mutaciones del gen del colágeno tipo I) afectan prácticamente a todos los huesos. de alguna manera los órganos del cuerpo humano . [15]
El colágeno tipo I está presente en todos los sistemas circulatorio y respiratorio : desde los ventrículos del corazón hasta las válvulas cardíacas y la vasculatura , [1] : 329 es una parte integral del tejido conectivo de los pulmones . [1] : 336 Como tal, las complicaciones cardiovasculares, entre ellas la insuficiencia aórtica , el aneurisma aórtico y las disecciones arteriales , a veces son comórbidas con la OI, [1] : 333 pero no con tanta frecuencia como lo son con el síndrome de Marfan . [1] : 332
Las enfermedades respiratorias son una de las principales causas de muerte en la OI. [12] [1] : 335 La fuente más obvia de problemas respiratorios en la OI es la insuficiencia pulmonar causada por problemas en la arquitectura de la pared torácica . [1] : 341 Sin embargo, las infecciones del tracto respiratorio , como la neumonía , también son más mortales entre las personas con OI que en la población general. [12] [39] Se encontró que aquellos con deformidades más graves de la caja torácica tenían una peor restricción pulmonar en un estudio a pequeña escala realizado en 2012 en el que participaron 22 pacientes italianos con OI tipos III y IV, además de 26 controles no afectados . [12]
La OI, especialmente su forma grave tipo III, también tiene efectos en el sistema gastrointestinal. Se encontró que estaba asociado con dolor abdominal recurrente y estreñimiento crónico en dos estudios en pacientes afectados por OI. [40] [41] El estreñimiento crónico es especialmente común, [1] : 377 y se cree que se ve agravado por una pelvis asimétrica ( protrusión acetabular ). [1] : 377 [41] Especialmente en la infancia, el estreñimiento asociado a la OI puede causar una sensación de saciedad y un rechazo de alimentos asociado, lo que lleva a la desnutrición . [1] : 377
Hay dos sistemas de tipificación para OI en uso moderno. El primero, creado por David Sillence en 1979, clasifica a los pacientes en cuatro tipos, o síndromes , según su presentación clínica , sin tener en cuenta la causa genética de su enfermedad. [42] : 114-115 [43] El segundo sistema amplía el modelo de Sillence, pero asigna genéticamente nuevos tipos numerados a medida que se encuentran. [44] [43] Por lo tanto, se puede describir que las personas con OI tienen tanto un tipo clínico como un tipo genético , que pueden ser equivalentes o no. [43]
El tipo I es el más común y el 90% de los casos son el resultado de mutaciones en COL1A1 o COL1A2 . [9] Los síntomas varían mucho entre tipos, así como también varían de persona a persona, incluso dentro de la misma familia. [45]
A partir de 2021, se han definido 21 tipos de IO: [16] [17]
Los cuatro tipos de Sillence tienen un significado tanto clínico como genético; Las descripciones a continuación son clínicas y se pueden aplicar a varios tipos genéticos de OI. Cuando se utiliza para referirse a un tipo genético y clínico, indica que los síntomas clínicos son causados por mutaciones en los genes COL1A1 o COL1A2 que se heredan de forma autosómica dominante. [dieciséis]
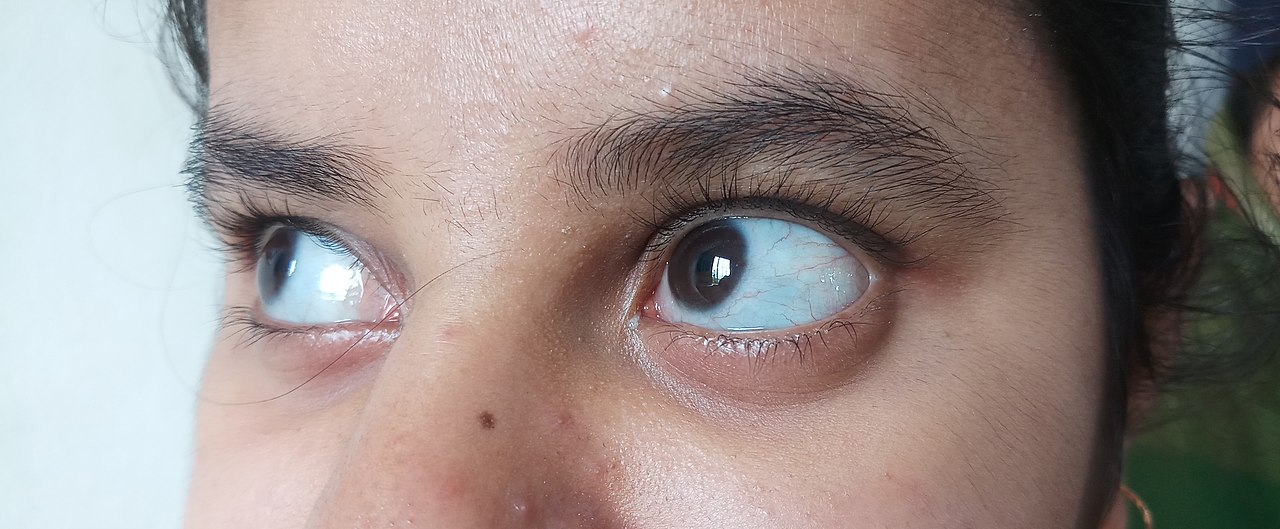
El colágeno es de calidad normal pero se produce en cantidades insuficientes. [5] : 1516 Los huesos se fracturan más fácilmente que en el público en general, pero no tan fácilmente como los tipos más graves de OI; puede haber escoliosis , aunque leve en comparación con la OI tipos III y IV, con un ángulo de Cobb más bajo ; las articulaciones pueden estar flojas; pueden ser evidentes escleróticas azules; es probable que se produzca pérdida de audición; [47] : Tabla 1 y podría haber una ligera disminución en la altura. Debido a que existen casos en los que faltan uno o más de estos síntomas, la OI tipo I en algunos casos no se detecta hasta la edad adulta. [5] : 1513-1514
Algunos dividen aún más el tipo I en tipos I – A y I – B, definidos por la ausencia (I – A) o presencia (I – B) de dentinogénesis imperfecta (dientes opalescentes). [47] [55] : 217 Las personas con tipo I generalmente tienen una esperanza de vida normal. [56]
El colágeno tiene un defecto fatal en su extremo C. [5] : 1512 La mayoría de los casos resultan en la muerte poco después del nacimiento, o dentro del primer año de vida, debido a insuficiencia respiratoria . Otra causa común de muerte son las hemorragias intracraneales por fracturas de cráneo presentes o sufridas durante o poco después del nacimiento. [5] : 1511 En muchos casos, el recién nacido ya tiene múltiples huesos rotos en el momento del nacimiento. Los bebés de tipo II también presentan graves problemas respiratorios y huesos gravemente deformados. El sesenta por ciento de los bebés mueren menos de 24 horas después de nacer y la supervivencia después del primer año es extremadamente improbable y normalmente requiere ventilación mecánica . [57] En los raros casos de bebés que sobreviven su primer año de vida, se observan retrasos motores y de desarrollo graves; Ninguno de los dos bebés estudiados en 2019, ambos de alrededor de dos años, había logrado el control de la cabeza y ambos necesitaban un ventilador para respirar. [58]
El tipo II también se conoce como la forma " perinatal letal " de OI, [59] y no es compatible con la supervivencia hasta la edad adulta. [57] Debido a huesos igualmente gravemente deformados, a veces los bebés con tipo III grave se clasifican inicialmente erróneamente como tipo II; una vez que se demuestra la supervivencia a largo plazo, se considera que tienen tipo III. [5] : 1511 [60]
La cantidad de colágeno es suficiente, pero no de calidad suficiente. [5] : 1512 La diferenciación clínica entre los tipos III y IV no siempre es sencilla y se complica aún más por el hecho de que un adulto con tipo IV no tratado puede tener peores síntomas que un adulto con tipo III tratado; [5] : 1511 [61] Las características que sólo se encuentran en el tipo III son su naturaleza progresivamente deformante [5] : 1511-1512 y la presencia de un rostro con apariencia "triangular". [62] Otro factor diferenciador entre el tipo III y IV son las escleróticas azules; en el tipo III, los bebés suelen tener escleróticas azules que gradualmente se vuelven blancas con la edad, pero las escleróticas azules no se observan comúnmente en el tipo IV, [1] : 294–296 , aunque se observan en el 10% de los casos. [63]
La OI tipo III provoca huesos osteopénicos que se fracturan con mucha facilidad, a veces incluso en el útero , lo que a menudo provoca cientos de fracturas a lo largo de la vida; [24] escoliosis temprana que progresa hasta la pubertad; enanismo (una altura adulta final frecuentemente inferior a 4 pies o 120 centímetros); articulaciones flojas; y posibles problemas respiratorios debido al bajo volumen de la caja torácica que causa volúmenes pulmonares bajos . [5] : 1512
Debido a la gravedad de los problemas óseos, es más probable que se desarrollen trastornos neurológicos y convulsivos en el tipo III. [5] : 1512 La invaginación basilar , que ejerce presión sobre el tronco del encéfalo , puede causar o contribuir a la muerte prematura; el tratamiento quirúrgico es más complejo en los casos de OI. [5] : 1512 [13] : 106–107
La cantidad de colágeno es suficiente, pero no de calidad suficiente. [5] : 1512 El tipo IV es para casos de gravedad variable, que no encajan ni en los tipos III ni en I. [47] Si bien una de las características requeridas por Sillence para el tipo IV era tener escleróticas normales, [1] : 294–296 [ 42] : 114 la clasificación moderna permite que incluso aquellos con escleróticas azules cumplan con los criterios del tipo IV si cumplen con los demás requisitos clínicos del tipo. [63]
En el tipo IV, la deformidad ósea puede ser de leve a grave, los huesos se fracturan fácilmente (especialmente antes de la pubertad), el enanismo es común, el colapso vertebral y la escoliosis son evidentes y es posible la pérdida de audición, [10] aunque es poco común. [47] [46] La IO tipo IV se define principalmente en contraste con el tipo III y el tipo I, siendo la clasificación clínica de los pacientes en algún punto intermedio entre los dos. [5] : 1511 Como tal, la IO tipo IV a menudo se denomina OI "variable", [42] : 111 y la gravedad incluso de aquellos en la misma familia (es decir, con la misma mutación genética) difiere. [47]
Las tasas de fractura ósea prepuberal son otra forma de evaluar clínicamente la OI tipo IV: quienes la padecen tienden a tener tasas de fractura de ≈1 por año, en comparación con ≈3 por año para la OI grave (tipo III). [47]
Al igual que en el tipo I, algunos dividen aún más el tipo IV en tipos IV-A y IV-B, definidos nuevamente por la ausencia (IV-A) o presencia (IV-B) de dentinogénesis imperfecta . [47] [55] : 217
A partir de 2020, quince tipos de OI se definen genéticamente: [16]
Dada la rápida tasa de descubrimiento de tipos, es muy probable que existan otros genes asociados con la OI que aún no se han informado. [83] : 491–492

La osteogénesis imperfecta es un grupo de trastornos genéticos, todos los cuales causan fragilidad ósea. La OI tiene una alta heterogeneidad genética , es decir, muchas mutaciones genéticas diferentes conducen a conjuntos de síntomas observables ( fenotipos ) iguales o similares. [84]
Las principales causas para desarrollar el trastorno son resultado de mutaciones en los genes COL1A1 y/o COL1A2 que son corresponsables de la producción de colágeno tipo I. [85] Aproximadamente el 90% de las personas con OI son heterocigotas para mutaciones en los genes COL1A1 o COL1A2 . [86] Hay varios factores biológicos que son resultados de la forma dominante de OI. Estos factores incluyen: estrés intracelular; mineralización anormal del tejido; interacciones anormales entre células; interacciones anormales entre células y matrices ; una estructura de matriz celular comprometida; e interacción anormal entre proteínas no colágenas y colágeno. [87]
Investigaciones anteriores llevaron a la creencia de que la OI era un trastorno autosómico dominante con pocas variaciones en los genomas. [8] Sin embargo, con la reducción del costo de la secuenciación del ADN a raíz del Proyecto Genoma Humano de 2003 , se han identificado formas autosómicas recesivas del trastorno. [88] Las formas recesivas de OI se relacionan en gran medida con defectos en las chaperonas de colágeno responsables de la producción de procolágeno y el ensamblaje de las proteínas relacionadas. [89] Ejemplos de chaperonas de colágeno que son defectuosas en pacientes con formas recesivas de OI incluyen la chaperona HSP47 ( síndrome de Cole-Carpenter ) y FKBP65 . [44] Las mutaciones en estas chaperonas dan como resultado un patrón de plegado inadecuado en las proteínas del colágeno 1 que causa la forma recesiva del trastorno. [44] Hay tres tipos importantes de OI que son el resultado de mutaciones en el complejo de colágeno prolil 3-hidroxilación (componentes CRTAP, P3H1 y CyPB). [44] Estos componentes son responsables de la modificación del colágeno α1(l)Pro986. [44] Las mutaciones en otros genes como SP7 , SERPINF1 , TMEM38B y BMP1 también pueden conducir a proteínas y enzimas formadas de forma irregular que dan como resultado otros tipos recesivo de osteogénesis imperfecta. [44]
Los defectos en las proteínas factor derivado del epitelio pigmentario (PEDF) y en la proteína transmembrana inducida por interferón restringida al hueso (BRIL) son las causas de la osteogénesis imperfecta tipo V y VI. [90] Los defectos en estas proteínas conducen a una mineralización ósea defectuosa , lo que provoca los huesos frágiles característicos de la osteogénesis imperfecta. [90] Una mutación puntual única en la región 5' no traducida (5' UTR) del gen IFITM5 , que codifica BRIL, está vinculada directamente a la OI tipo V. [64] [91]
En el raro caso del tipo XIX, descubierto por primera vez en 2016, la OI se hereda como un trastorno genético ligado al cromosoma X , y sus efectos perjudiciales resultan en última instancia de una mutación en el gen MBTPS2 . [81] La investigación genética está en curso y no está claro cuándo se identificarán todas las causas genéticas de la OI, ya que la cantidad de genes que deben analizarse para descartar el trastorno continúa aumentando. [83] : 491–492
En un estudio de 37 familias, se encontró una probabilidad del 1,3% de que la OI reaparezca en varios hermanos nacidos de dos padres no afectados; esta es una tasa mucho más alta de lo que se esperaría si todas esas recurrencias fueran de novo . [92] La causa es el mosaicismo genético ; es decir, algunas o la mayoría de las células germinales de uno de los padres tienen una forma dominante de OI, pero no suficientes células somáticas como para causar síntomas o una discapacidad obvia en el padre; las diferentes células del padre tienen dos (o más). ) conjuntos de ADN ligeramente diferentes. [92] [5] : 1513 Se ha observado clínicamente que ≈5 a 10% de los casos de OI tipos II y III son atribuibles al mosaicismo genético. [1] : 532
Las personas con OI nacen con tejido conectivo defectuoso , sin la capacidad de producirlo en cantidades suficientes o, en los tipos genéticos más raros, nacen con deficiencias en otros aspectos de la formación ósea , como las proteínas chaperonas , la vía de señalización Wnt , la proteína BRIL , etcétera. [16] En el tipo I, la estructura del colágeno en sí es normal, lo único que es bajo es su cantidad. [5] : 1516 Los tipos II, III y IV suelen estar relacionados, aunque no siempre, con una deficiencia de colágeno tipo I. [93] Una posible deficiencia surge de una sustitución de un aminoácido de glicina por un aminoácido más voluminoso, como la alanina , en la estructura de triple hélice de la proteína de colágeno . Las cadenas laterales de aminoácidos más grandes provocan efectos estéricos que crean un abultamiento en el complejo de colágeno, lo que a su vez influye tanto en la nanomecánica molecular como en la interacción entre moléculas , ambas comprometidas. [94] [95] Dependiendo tanto de la ubicación de la sustitución como del aminoácido que se utiliza en su lugar, se observan diferentes efectos que explican la diversidad de tipos en la OI a pesar de que los mismos dos genes de colágeno son responsables de la mayoría de los casos. [96] [95] Los reemplazos de glicina con serina o cisteína se observan con menos frecuencia en la IO fatal tipo II, mientras que los reemplazos con valina , ácido aspártico , ácido glutámico o arginina se observan con mayor frecuencia. [95]
A mayor escala, la relación entre las fibrillas de colágeno y los cristales de hidroxiapatita para formar hueso se altera, provocando fragilidad. [96] Las fracturas óseas se producen porque el estado de tensión dentro de las fibrillas de colágeno se altera en los lugares de las mutaciones, donde fuerzas de corte localmente mayores conducen a una falla rápida de las fibrillas incluso con cargas moderadas porque se pierde el estado de tensión homogéneo que normalmente se encuentra en las fibrillas de colágeno sanas. [94] La OI es, por lo tanto, un fenómeno de múltiples escalas, donde los defectos en los niveles más pequeños de los tejidos (genéticos, nano, micro) se dominó para afectar el nivel macro de los tejidos . [94]
El diagnóstico generalmente se basa en imágenes médicas , incluidas radiografías simples , y en los síntomas. En la OI grave, los signos en las imágenes médicas incluyen anomalías en todas las extremidades y en la columna. [97] Como los rayos X a menudo son insensibles a la pérdida de densidad ósea comparativamente menor asociada con la OI tipo I, es posible que se necesiten exploraciones DEXA . [5] : 1514
Un diagnóstico de OI se puede confirmar mediante análisis de ADN o de proteínas de colágeno , pero en muchos casos, la aparición de fracturas óseas con poco trauma y la presencia de otras características clínicas como escleróticas azules son suficientes para un diagnóstico. Se puede realizar una biopsia de piel para determinar la estructura y cantidad de colágeno tipo I. Si bien las pruebas de ADN pueden confirmar el diagnóstico, no pueden excluirlo por completo porque aún no se conocen ni se analizan todas las mutaciones que causan la OI. [83] : 491–492 La OI tipo II a menudo se diagnostica mediante ecografía durante el embarazo, donde ya pueden ser visibles múltiples fracturas y otros rasgos característicos. En relación con el control, el hueso cortical OI muestra mayor porosidad, diámetro del canal y conectividad en la tomografía microcomputarizada. [98] La IO también se puede detectar antes del nacimiento mediante el uso de una técnica de prueba genética in vitro como la amniocentresis . [99]
Para determinar si hay osteogénesis imperfecta, se puede realizar la secuenciación genética de los genes problemáticos más comunes, COL1A1 , COL1A2 e IFITM5 ; [100] Si no se encuentra ninguna mutación y todavía se sospecha OI, se pueden analizar los otros 10+ genes que se sabe que causan OI. [16] También se sugiere realizar pruebas de duplicación y eliminación a los padres que sospechan que su hijo tiene OI. [100] La presencia de mutaciones de cambio de marco causadas por duplicaciones y eliminaciones es generalmente la causa de una mayor gravedad de la enfermedad. [100]
Un diagnóstico diferencial importante de OI es el abuso infantil , ya que ambos pueden presentarse al médico con múltiples fracturas en diversas etapas de curación. [5] : 1514 Diferenciarlos puede ser difícil, especialmente cuando no están presentes otros rasgos característicos de la OI. [1] : 391 Esto puede convertirse en un problema en los tribunales; En los Estados Unidos, varios casos de abuso infantil se resolvieron con el hallazgo de que la osteogénesis imperfecta era la verdadera causa de las fracturas de un niño, lo que dio lugar a demandas que buscaban reparación, como la de Alice Velasquez, et al. contra Estados Unidos . [1] : 391 [101]
Otros diagnósticos diferenciales incluyen el raquitismo y la osteomalacia , ambos causados por desnutrición , así como síndromes esqueléticos raros como el síndrome de Bruck , la hipofosfatasia , el geroderma osteodisplásico y el síndrome de Ehlers-Danlos . [5] : 1513 [1] : 253–256 Varias formas de osteoporosis , como la osteoporosis iatrogénica , la osteoporosis juvenil idiopática , la osteoporosis por desuso y la osteoporosis relacionada con el ejercicio, también deben considerarse como explicaciones cuando se sospecha OI. [1] : 255–256
No existe cura para la osteogénesis imperfecta. [10] Mantener un estilo de vida saludable haciendo ejercicio y evitando fumar puede ayudar a prevenir fracturas. [102] El tratamiento puede incluir atención de huesos rotos, analgésicos, fisioterapia , ayudas para la movilidad, como aparatos ortopédicos o sillas de ruedas, y cirugía. [102]
Juzgar el éxito o el fracaso del tratamiento puede resultar difícil en pacientes con OI, ya que la disminución de las tasas de fracturas óseas puede ser simplemente una coincidencia. Si bien estas tasas se utilizan a menudo en estudios médicos para juzgar la eficacia del tratamiento, un estudio noruego de quince personas con OI enfatizó que sienten que los médicos deberían considerar al paciente en su totalidad y no solo las tasas de fracturas. [103]
Las fracturas óseas se tratan en personas con osteogénesis imperfecta de la misma manera que se tratan en la población general: el hueso OI sana al mismo ritmo que el hueso no OI. [1] : 431 Se pone mayor énfasis en el uso de materiales livianos para inmovilizar la fractura, ya que en los tipos de OI moderada o grave, el uso de yesos pesados , como los yesos en espica de cadera , puede causar fracturas en los huesos en los límites del yeso. , así como osteopenia generalizada . [1] : 431 El yeso o férula liviano se reemplaza con una ortesis removible después de algunas semanas y una vez que se observa evidencia de consolidación en la radiografía. [1] : 431 Para prevenir una pseudoartrosis o una consolidación defectuosa , todas las fracturas deben inmovilizarse, incluso si la fractura parece trivial ( microfractura ), [1] : 439 ya que las personas con OI tienen un mayor riesgo de pseudoartrosis. [1] : 438
Las infecciones óseas secundarias a fracturas se tratan a medida que se presentan con los antibióticos y antisépticos adecuados , al igual que en la población general. [1] : 424

En 1998, un ensayo observacional inicial demostró la eficacia del pamidronato intravenoso , un bifosfonato que se había utilizado previamente en adultos para tratar la osteoporosis . En la OI grave, este ensayo demostró que el pamidronato redujo el dolor óseo, previno nuevas fracturas vertebrales, reformó los cuerpos vertebrales previamente fracturados y redujo el número de fracturas de huesos largos. [104]
Aunque los bifosfonatos orales son más convenientes y baratos, no se absorben tan bien y los bifosfonatos intravenosos son generalmente más eficaces, aunque esto está en estudio. Algunos estudios han encontrado equivalentes los bifosfonatos orales e intravenosos, como el alendronato oral y el pamidronato intravenoso. [105] En un ensayo doble ciego realizado en 2013 en niños con OI leve, el risedronato oral aumentó la densidad mineral ósea y redujo las fracturas no vertebrales. Sin embargo, no disminuyó las nuevas fracturas vertebrales. [106] [107] Una revisión Cochrane de 2016 concluyó que, aunque los bifosfonatos parecen mejorar la densidad mineral ósea, no está claro si esto conduce a una reducción de las fracturas óseas o a una mejora en la calidad de vida de las personas con osteogénesis imperfecta. [7] Incluso en ensayos con hasta 125 niños, no se ha encontrado ningún vínculo causal entre los bifosfonatos y la disminución de las tasas de fracturas; Los ensayos controlados con placebo tampoco pudieron demostrar que provocaran un aumento de la fuerza, el control motor o niveles más bajos de dolor. [87]
Los bifosfonatos no son tan eficaces para aumentar la densidad mineral ósea en los adultos. [22]
La OI es un trastorno genético y no es causada por una ingesta insuficiente de ninguna vitamina o mineral; los suplementos no pueden curar la OI. Sin embargo, las personas con OI tienden a tener una deficiencia grave de vitamina D en tasas mucho más altas que la población general, y la causa de esto no se comprende bien. [108] [109] [110] Se cree que la gravedad de la deficiencia y la probabilidad de que ocurra están relacionadas con la gravedad de la OI. [109] Se puede recomendar la suplementación con vitamina D, al menos hasta que los niveles de 25(OH)D 3 en la sangre del paciente vuelvan a la normalidad. [108] La deficiencia de vitamina D también es una preocupación, ya que puede disminuir el beneficio de los bifosfanatos. [108]
Una cirugía de cualquier tipo conlleva inherentemente más riesgos cuando se realiza en un paciente que tiene OI (especialmente de moderada a grave). Las deformidades esqueléticas y la dentinogénesis imperfecta pueden dificultar el acceso a las vías respiratorias . [1] : 333 El uso y la retirada de la ventilación mecánica también son más difíciles de llevar a cabo en pacientes con OI. [1] : 333 Durante el procedimiento en sí o el proceso de curación, el colágeno defectuoso de la OI puede provocar diátesis hemorrágicas . [1] : 333
La seguridad de la anestesia también es motivo de mayor preocupación entre los pacientes con OI, [1] : 333 con complicaciones anestésicas 5,6 veces más probables que ocurran cuando el paciente tiene OI tipo III. [111] Una preocupación única de la anestesia en la OI es la fractura perioperatoria : fracturas sufridas debido al traslado del paciente y a las técnicas de acceso a las vías respiratorias que, si bien son rutinarias cuando los huesos de un paciente son fuertes, pueden causar lesiones con huesos frágiles en la OI. [112] A modo de ejemplo, debido a un informe de 1972 sobre una fractura de húmero causada por el manguito de un esfigmomanómetro sostenida en un paciente con OI durante una cirugía, los protocolos de monitoreo de la presión arterial a menudo se modifican para pacientes con OI, y se utilizan manguitos de tamaño neonatal y ajustes de la máquina incluso en adultos; [113] : ¶11.72 además, se prefiere que las extremidades menos deformadas del paciente reciban el manguito. [20] : ¶14.23
Se pueden insertar quirúrgicamente varillas de metal en los huesos largos para mejorar la fuerza, un procedimiento desarrollado por Harold A. Sofield cuando era jefe de personal de los Hospitales Shriners para Niños de Chicago , un hospital que ofrece atención ortopédica y cirugía a niños independientemente de la capacidad de su familia. pagar. [115] Un gran número de niños con OI acudieron a Shriners y Sofield experimentó con varios métodos para fortalecer sus huesos. [116] En 1959, con Edward A. Millar [ sic ], Sofield escribió un artículo fundamental que describía una cirugía de tres partes que parecía radical en ese momento: romper con precisión los huesos ("fragmentación"), colocar los fragmentos óseos resultantes en un línea recta ("realineación") y luego colocar varillas metálicas en los canales intramedulares de los huesos largos para estabilizarlos y fortalecerlos ("fijación con varillas"). [117] Su tratamiento resultó útil para aumentar la movilidad de las personas con OI y ha sido adoptado en todo el mundo; se convirtió en el tratamiento quirúrgico estándar para la OI grave en 1979, año en el que David Sillence descubrió que ≈ 2 ⁄ 3 de los pacientes El encuestado con OI tipo III se había sometido al menos a una cirugía de varilla. [42] : 108
La cirugía con Rodding a menudo se realiza con la esperanza de ofrecer un camino hacia la deambulación, caminar , a pacientes con OI moderada o grave. Una revisión de 2020 en The Journal of Bone and Joint Surgery ( JB&JS ) encontró que sigue siendo muy popular: ≈ 2 ⁄ 3 de las personas con OI tipos III y IV (OI grave) se han sometido a algún tipo de cirugía con varilla en sus vidas, en un promedio edad de 4+1 ⁄ 10 y 7+1 ⁄ 2 años respectivamente; [25] : Tabla I Una posible explicación para una tendencia hacia una intervención más temprana en el tipo III es que la mitad de los niños afectados no podían caminar en absoluto sin la cirugía, ya que sus extremidades estaban más arqueadas, por lo que se buscó la cirugía antes. [25]
En aquellos con OI tipo III que se habían sometido a una cirugía con varillas, al 79,5% se les colocaron varillas en los fémures y las tibias de ambas piernas. [25] : Tabla I La forma más común de varillas utilizadas son las varillas intramedulares (IM) , algunas de las cuales, como la varilla IM de Fassier-Duval, son telescópicas , lo que significa que están diseñadas para crecer a medida que crece el niño, en un intentar evitar la necesidad de cirugías de revisión. [118] Las varillas IM telescópicas se utilizan ampliamente, [119] y la varilla IM común de Fassier-Duval está diseñada para usarse con varillas en el fémur, la tibia y el húmero. [120] : 1 La cirugía implica romper los huesos largos entre uno y tres (o más) [119] : Figura 4 lugares, y luego fijar la varilla a lo largo del hueso para mantenerlo recto. [120] : 11
Mientras que las varillas telescópicas IM están diseñadas para crecer junto con el fémur y la tibia en los niños en desarrollo; los cirujanos tienen preferencia por utilizar varillas IM no telescópicas, como las varillas Rush, en la tibia, que crece menos comparativamente; la revisión de JB&JS encontró que, si bien el 69,7 % de los fémures fueron tratados con varillas IM telescópicas, solo el 36,9 % de las tibias lo fueron. [25] : Tabla IV
Si bien la revisión en JB&JS pudo correlacionar la cirugía con varillas con una mayor movilidad en todos los tipos de OI, en pacientes con tipo IV, la cirugía no disminuyó la incidencia de huesos rotos en comparación con los pacientes sin varillas, mientras que los pacientes con tipo IV con tibias con varilla experimentaron 0,93 fracturas de tibia por año, los pacientes con tibias naturales experimentaron solo 0,81. Sin embargo, en pacientes con tipo III, la cirugía con varilla disminuyó el número promedio de fracturas de tibia por año de 0,84 a 0,57. [25] : Tabla V
La fusión espinal se puede realizar como medida preventiva o para corregir la escoliosis existente , aunque la fragilidad inherente del hueso con OI hace que esta operación sea más compleja en pacientes con OI que en pacientes con escoliosis idiopática adolescente , pero densidad ósea normal. [121] Sin embargo, a pesar de los riesgos, tres cirujanos ortopédicos de Nemours-duPont que se especializan en intervenciones quirúrgicas para la osteogénesis imperfecta recomiendan operar si la curva es mayor a 50° después de que el niño haya superado la velocidad máxima de altura , ya que la curva de la columna puede continuar empeorando. incluso hasta la edad adulta. [13] : 104
Debido al riesgo que implica, los mismos cirujanos recomiendan que la cirugía de impresiones e invaginaciones basilares sólo se realice si la presión que se ejerce sobre la médula espinal y el tronco del encéfalo está provocando síntomas neurológicos reales. [13] : 106–107 Una vez que la invaginación basilar se ha vuelto sintomática, sólo la cirugía puede detener o revertir la progresión de los déficits neurológicos . [1] : 345
Generalmente se recomienda la fisioterapia , aunque se requieren protocolos individualizados debido a la variabilidad de la IO. [1] : 378 La fisioterapia se utiliza para fortalecer los músculos, mejorar la motilidad , mejorar la flexibilidad y ayudar con el mantenimiento del peso , aunque debe realizarse de manera suave para minimizar el riesgo de fractura ósea. [1] : 378 En personas con OI, el ejercicio a menudo incluye ejercicios aeróbicos acuáticos , ejercicios ligeros de resistencia y caminar, si el paciente puede. [1] : 378 Sin embargo, incluso en pacientes con OI leve, los deportes de contacto , así como las actividades que puedan ejercer una tensión innecesaria en las articulaciones, como saltar , están contraindicados debido a los riesgos que plantean. [1] : 378
Se anima a las personas con movilidad más limitada a cambiar de posición regularmente a lo largo del día; A las personas que se sientan en una silla de ruedas la mayor parte o todo el día se les recomienda salir de ella cada dos horas, como forma de ejercicio, para disminuir la rigidez y prevenir las úlceras por presión . [1] : 378
Las personas con OI de moderada a grave, que requieren dispositivos de asistencia a la movilidad y vehículos adaptados , enfrentan barreras importantes para acceder a piscinas o gimnasios accesibles para sillas de ruedas ; es posible que no tengan ninguno en su área ni los medios para llegar allí. [1] : 378 Es más probable que la obesidad se presente entre personas con OI grave (especialmente después de los 20 años) y, en algunos, puede causar una mayor disminución de la movilidad. [122] [1] : 371, 373
También se puede realizar vibración de todo el cuerpo con mesa inclinada para aumentar la movilidad de pacientes con OI inmovilizados a largo plazo ( postrados en cama ); en al menos dos casos ayudó a los niños postrados en cama a poder sentarse erguidos. [123] [87]
Más de 1 de cada 2 personas con OI también tiene dentinogénesis imperfecta (DI), una anomalía congénita en la formación de la dentina , uno de los cuatro componentes principales del diente humano . [124] El tratamiento dental puede representar un desafío como resultado de las diversas deformidades, esqueléticas y dentales, debido a la OI. Los niños con OI deben acudir a un chequeo dental tan pronto como les salgan los dientes ; esto puede minimizar la pérdida de estructura dental como resultado de una dentina anormal, y deben ser monitoreados regularmente para preservar sus dientes y su salud bucal. [124]
Muchas personas con OI reciben tratamiento con bifosfonatos y existen varias posibles complicaciones relacionadas con los procedimientos dentales, por ejemplo, osteonecrosis de la mandíbula relacionada con medicamentos (MRONJ). Sin embargo, en una revisión Cochrane de 2016 sobre la seguridad y eficacia de los bifosfonatos para la OI no se encontró ningún informe de MRONJ relacionado con los bifosfonatos en un niño o un adulto con OI . [7]
Los anticuerpos monoclonales se han considerado durante mucho tiempo para la OI, pero hasta 2021, dicha terapia no ha sido aprobada para la OI, ni en la Unión Europea ni en los Estados Unidos. Por tanto, no está claro si son seguros o eficaces. Entre los anticuerpos monoclonales que se han estudiado se encuentran romosozumab (se dirige a la esclerostina , de Amgen ), fresolimumab ( TGF-β , Sanofi ), blosozumab (esclerostina, Lilly ) y setrusumab (esclerostina, iniciado por Novartis ). [125]
Setrusumab, anteriormente conocido como BPS-804, es un anticuerpo monoclonal que se dirige a la esclerostina y se ha estudiado específicamente en la OI más que cualquier otro. En el cuerpo, la esclerostina se une a los receptores LRP5 y LRP6 , lo que inhibe la vía de señalización Wnt . Esto disminuye la formación de hueso y no es un problema cuando una persona tiene huesos sanos. [126] Sin embargo, se cree que disminuir la concentración de esclerostina en el cuerpo puede conducir a la formación de más hueso, y esa es la premisa de por qué los anticuerpos monoclonales que reducen las concentraciones de esclerostina natural pueden ayudar a fortalecer el hueso OI. . [127] Si bien setrusumab se desarrolló por primera vez en la compañía farmacéutica Novartis, Novartis vendió sus derechos para patentar el medicamento a Mereo Biopharma en 2015, quien continuó su desarrollo junto con Ultragenyx . [125] [128] En 2019, Mereo anunció que había concluido la recopilación de datos para su ensayo de fase II-B de setrusumab; el estudio se completó el 12 de noviembre de 2020. [125] [129] A pesar de que los datos del ensayo no lograron mostrar mejoras en la densidad ósea en las exploraciones QCT , su objetivo principal, hubo mejoras en las exploraciones DXA . [130] En un comunicado de prensa de septiembre de 2020, Mereo dijo que estaba buscando realizar un ensayo de fase III en 2021 y que había recibido una designación de Enfermedad Pediátrica Rara (RPD) de la Administración de Medicamentos y Alimentos de EE. UU. (FDA). [131]
Romosozumab , que también es un anticuerpo monoclonal dirigido a la esclerostina, es un fármaco aprobado en EE. UU. y la UE para el tratamiento de la osteoporosis . El analista de la industria farmacéutica Evercore ha señalado que "podría acabar con la economía de setrusumab ", ya que romosozumab tiene un precio más barato que el de un medicamento para una enfermedad rara, afirmando que será "vital" para los márgenes de beneficio de Ultragenyx demostrar que su setrusumab es más eficaz que romosozumab para la OI. [125] Un ensayo clínico que evalúa la eficacia de romosozumab en la OI comenzó en septiembre de 2020 y a partir de septiembre de 2021 está en curso. [132] Ultragenyx predice que sus ensayos de fase 2/3 para setrusumab se completarán en 2026. [133] [134]

Como trastorno genético, el pilar de la prevención de la osteogénesis imperfecta en el siglo XXI se basa en evitar que los individuos afectados nazcan en primer lugar . El asesoramiento genético puede ayudar a los pacientes y sus familias a determinar qué tipos de pruebas de detección, si las hay, son adecuadas para su situación. Los pacientes pueden considerar el diagnóstico genético preimplantacional después de la fertilización in vitro para seleccionar embriones fertilizados que no se vean afectados. [1] : 247–248 Las mutaciones comunes que causan OI pueden detectarse mediante la secuenciación del exoma y la secuenciación del genoma completo . Si ya hay un embarazo en curso, se puede realizar el procedimiento de amniocentesis para ver si el feto está afectado. [1] : 247 Si se ve afectado, corresponde a la familia considerar si desea o no interrumpir el embarazo y volver a intentarlo, lo que plantea cuestiones de ética médica y el derecho de la mujer a elegir . [135] [136]
Sin intervención, los pacientes con las mutaciones más comunes que causan osteogénesis imperfecta tienen un 50% de posibilidades por gestación de transmitir el trastorno, ya que estas mutaciones se heredan con un patrón autosómico dominante de herencia mendeliana . [1] : 247 Aquellos con formas raras autosómicas recesivas de OI tienen un 25 % de posibilidades de transmitir el trastorno. Se pueden utilizar pruebas genéticas de los miembros afectados de la familia para determinar qué patrón de herencia se aplica. [1] : 101
Como la OI tipo I puede ser difícil de detectar en un recién nacido, se puede analizar la sangre del cordón umbilical del niño para determinar si se ha transmitido si la familia ya ha rechazado los métodos de detección genética más invasivos. [1] : 247 En casos más graves, el diagnóstico se puede realizar mediante ecografía, especialmente si la IO ya es una posibilidad. [1] : 248 Una preocupación ética con la detección prenatal de OI surge a menudo cuando los padres preguntan qué tan gravemente afectado estará su hijo; estas preguntas aún son difíciles de responder de manera concluyente. [1] : 382
Si una persona no afectada ya ha tenido un hijo con OI, existe una mayor probabilidad (aunque todavía bastante remota) de que sus futuros hijos tengan OI debido al mosaicismo genético . [92] [1] : 100, 1513
La crítica de los derechos de las personas con discapacidad a la detección prenatal de IO, sostenida por algunos especialistas en bioética y algunas personas afectadas, la compara negativamente con la eugenesia , e incluso aquellos que no se oponen al aborto se oponen a los abortos selectivos con el argumento ético de que su existencia traiciona la creencia de que las vidas de aquellos con OI son "menos dignos de vivir [y] menos valiosos". [1] : 388
El pronóstico de la osteogénesis imperfecta depende completamente de su tipo (ver § Clasificación).
En la forma leve del trastorno, tipo I, la esperanza de vida de los pacientes es cercana a la de la población general. [1] : 461 Sin embargo, en el tipo II, los pacientes rara vez viven más de dos años y generalmente mueren en sus primeras semanas de vida. [5] : 1511 La evaluación de la esperanza de vida de los pacientes con tipos III y IV es más complicada, ya que las elecciones de estilo de vida pueden causar lesiones traumáticas mortales que de otro modo no habrían ocurrido o no habrían sido mortales en la población general. Se cree que la esperanza de vida en la IO tipo IV es cercana a la normal, pero en la tipo III es menor que en la población general. [48]
de Dinamarca encontró que en todos los tipos de IO, la mortalidad por todas las causas era tres veces mayor, lo que llevaba a una pérdida de alrededor de siete años en las mujeres y nueve años en los hombres. [15] Un estudio de 1996 publicado en el British Medical Journal encontró que la mortalidad en la IO tipo III es significativamente mayor, y muchos pacientes mueren entre los 20, 30 y 40 años; Además, se descubrió que los pacientes que sobreviven hasta los 10 años tienen una esperanza de vida más larga que los recién nacidos. [137]
Las personas con OI leve (tipo I) en la edad adulta necesitan pocos equipos de adaptación , aunque en la infancia alcanzan los hitos motores con un retraso significativo en comparación con la población general. [1] : 477
Con equipos de adaptación como muletas , sillas de ruedas motorizadas , férulas , extensores de alcance y/o modificaciones en el hogar, muchas personas con OI de moderada a grave pueden lograr o mantener un grado significativo de independencia . [18] [1] : 488 Con tratamiento y fisioterapia, se espera que los niveles máximos de movilidad sean caminata comunitaria sin asistencia para el tipo I, caminata doméstica o de ejercicio para el tipo III y caminata doméstica o comunitaria para el tipo IV; Debido a la variabilidad de la OI entre individuos, la movilidad lograda varía y puede estar por debajo de este máximo esperado. [1] : 476
En Estados Unidos, se estima que la incidencia de osteogénesis imperfecta es de uno por cada 20.000 nacidos vivos. [138] Se estima que entre 20.000 y 50.000 personas se ven afectadas por la IO en los Estados Unidos. [139]
Los tipos más comunes son I, II, III y IV, mientras que el resto son muy raros. [140] El tipo I es el más común y se ha informado que es aproximadamente tres veces más común que el tipo II. La prevalencia de los tipos III y IV es menos segura. [23] En un estudio de 1989 en Dinamarca, se encontró que el tipo I comprendía el 71% de los casos y el tipo II el 12% de los casos, mientras que otros tipos comprendían el 17% restante. [15] En un estudio realizado en Suecia en 2015, el tipo I fue casi seis veces más común que el tipo III y casi cuatro veces más común que el tipo IV. [24]
La mayoría de las personas con OI la reciben de uno de sus padres, pero en muchos casos se trata de una mutación nueva ( de novo o "esporádica") en una familia. En un estudio de pacientes con tipos de OI que pueden sobrevivir, la OI tipo III es más frecuente de novo (85%), seguida del tipo IV (50%) y el tipo I (34%). [6] : Tabla 1
Algunas poblaciones pueden tener una incidencia de OI mayor de lo que se esperaría si tienen un número mayor que el promedio de portadores de las formas recesivas de la enfermedad. [1] : 20–21 [141]
La afección, o sus tipos, ha tenido otros nombres a lo largo de los años y en diferentes países; Sin embargo, "osteogénesis imperfecta" ha sido el nombre más aceptado para esta afección desde finales del siglo XX. Entre algunas de las alternativas más comunes se encuentran la “fragilitas ossium”; [2] "Síndrome de Ekman-Lobstein" y "síndrome de Vrolik", ambos epónimos ; y, el coloquialismo , "enfermedad de los huesos de cristal". [1]
OI ha sido identificado en un niño del antiguo Egipto momificado alrededor del año 1000 a. C., originalmente descartado por los arqueólogos por contener restos de un mono . [142] [143] : 161 El rey nórdico Ivar el Deshuesado , que vivió c. 800 CE , [144] se especula que también tuvo OI. [145]
A Nicolas de Malebranche se le atribuye a menudo el mérito de ser la primera persona en describir las características físicas de OI en su libro de 1688 La búsqueda de la verdad , en el que describe a un hombre al que le han roto "huesos en los lugares en los que estaría un asesino". " toda su vida. Sin embargo , su confiada descripción de la patología del trastorno, que crea lo que denominó « enfants monstrueux » ("niños monstruosos"), es científicamente vacía: escribió que se debió a que la madre vio antes del parto una ejecución pública al romper una rueda. . [27] : 683 [143] : 165–168 [146]
Los primeros estudios científicos modernos sobre la OI comenzaron en 1788 por Olof Jakob Ekman, quien describió la enfermedad, que denominó " osteomalacia congénita", en su tesis doctoral y mencionó casos de la misma que se remontaban a 1678, todos en la misma familia, a través de tres generaciones. [147] La descripción de Ekman de la afección mencionaba enanismo, fragilidad ósea e inclinación de los huesos largos. [148] : 763 En 1831, Edmund Axmann dio una descripción detallada de la misma en él y en sus dos hermanos, siendo el primero en mencionar las escleróticas azules como un signo característico de la OI. [27] : 683 Jean Lobstein describió por primera vez la forma leve de la enfermedad, hoy conocida como tipo I, en 1833, llamándola "osteopsatirosis idiopática ". [3] : 347
No fue hasta 1912 que la pérdida de audición se reconoció positivamente como un síntoma de OI, mencionado por primera vez en un breve artículo del médico inglés Charles Allen Adair-Dighton. [149] [143] : 168–169
Willem Vrolik , un anatomista holandés que también fue curador del "Museum Vrolikianum", lo que le permitió conocer muchos especímenes de cuerpos con defectos de nacimiento , acuñó el término "osteogénesis imperfecta" [27] : 683 en su libro bilingüe en latín y holandés. sobre teratología , Ilustraciones de embriogénesis humana y de mamíferos , publicado por primera vez en 1849. [150]
Se incluye una descripción de los restos de un bebé que tenía lo que ahora se conoce como OI perinatalmente fatal tipo II [3] : 347 [1] : 5 (como se verificó en un nuevo examen de los restos realizado en 1998 por Baljet et al. ) . [151] Los restos fueron entregados primero al padre de Vrolik, quien no podía entenderlos. Vrolik describió huesos pobremente mineralizados, huesos largos arqueados y fracturas en diversos estados de curación. [152] Vrolik determinó correctamente que lo que denominó OI en el lactante no era causado por raquitismo secundario , sino por una anomalía congénita que causaba osteopenia primaria ; Teorizó que esto se debía a una falta de "energía generativa intrínseca". [151]
La clasificación de la IO también ha evolucionado a medida que ha mejorado su comprensión científica. Antes de la llegada de las pruebas genéticas modernas, la OI se clasificaba en dos grandes grupos: osteogénesis imperfecta congénita y osteogénesis imperfecta tarda , una división propuesta por primera vez por el médico alemán E. Looser en 1906. [2] [153] Congénita se utilizó para describir los tipos clínicos modernos II, III y algunos casos de IV, donde al nacer el padecimiento era evidente, ya sea por arqueamiento de los miembros o por fracturas sufridas en el útero . [42] Tarda se utilizó para clasificar la OI moderna tipo I y algunos casos de tipo IV, donde la fragilidad inherente de los huesos no quedó clara hasta mucho después del nacimiento. [2] La idea de que estas formas "tardías" y "prenatales" eran manifestaciones del mismo trastorno fue propuesta por primera vez en 1897 por Martin Benno Schmidt ; [154] en la década de 1950 este hecho era bien aceptado. [3] : 346
Mientras tanto, el sistema moderno de cuatro tipos (I, II, III, IV) fue introducido en un artículo de David Sillence , Alison Senn y David Danks en el Journal of Medical Genetics en 1979, [42] [155] y tiene desde entonces se han convertido en términos estándar entre médicos, pacientes e investigadores. [43] [47] [156] Los tipos genéticos modernos (aquellos con números mayores que IV) se han utilizado a medida que se han descubierto formas cada vez más recesivas de OI desde el descubrimiento del primero por Roy Morello et Alabama. en 2006. [46] [8] [67] En 2010, el Grupo Internacional de Nomenclatura para los Trastornos Constitucionales del Esqueleto (INCDS) "liberó" los tipos Sillence de la referencia molecular , aceptando su nuevo papel clínico como prioridad a raíz de lo que para ellos fue un aumento "sorprendente" en el número de causas genéticas de la OI. [156] [47]
En un artículo para la Revisión Anual de Genética de 2012, los Dres. Peter Byers y Shawna M. Pyott lamentaron cómo la expansión del número de tipos para incluir tipos genéticos ha creado un sistema que " creció como Topsy ". [83] : 492 Sugieren que, de hecho, puede ser imposible crear un sistema que sea útil para los médicos y que describa con precisión la causa genética de la OI de una persona, con intentos siempre priorizando un uso a expensas del otro. [83] : 492
Se han realizado muchas investigaciones médicas sobre las causas de la osteogénesis imperfecta, beneficiando no sólo a las personas con OI sino también a la medicina en general; En los diez años transcurridos entre 2006 y 2016, los numerosos descubrimientos de mutaciones genéticas recesivas no relacionadas con el colágeno, que aún conducen a los signos clínicos de OI en quienes las padecen, condujeron a numerosos avances en la comprensión médica del proceso de desarrollo óseo sano. . [8]
En los perros, la OI es una afección autosómica recesiva , lo que significa que los perros con dos copias del alelo se verán afectados. [157] Muchas organizaciones de raza y veterinarios ofrecen pruebas de OI para saber si un perro es portador de OI. [157] [158] Para prevenir la OI, los perros que son heterocigotos para la OI solo deben cruzarse con no portadores. [158]
Se han encontrado mutaciones naturales que causan OI en Golden Retrievers , Dachshunds y Beagles . También se ha identificado OI en peces cebra y ratones . [159]
Aunque los perros, ratones, peces y humanos no son genéticamente idénticos, se ha reconocido oficialmente que algunos de estos modelos animales representan los distintos tipos de OI en humanos. Por ejemplo, los ratones homocigotos oim / oim experimentan fracturas óseas espontáneas, tamaño corporal pequeño y cifosis, lo que los convierte en un modelo de OI tipo III. Mientras tanto, los ratones heterocigotos oim /+ parecen normales pero tienen huesos que son bastante más débiles que los ratones salvajes, lo que los convierte en un modelo para la OI tipo I. [159] [160] Al igual que en la OI humana, la ubicación en el gen que está mutado afecta la gravedad de la enfermedad resultante: el ratón G859C Col1a1 es un modelo para la OI tipo II, ya que todos los ratones afectados mueren en el período perinatal. [159]
Las pruebas con animales en modelos animales identificados pueden conducir a terapias humanas para la OI. [159]
La dentinogénesis imperfecta (DI) es más frecuente en los tipos III y IV de OI y, en general, afecta aproximadamente al 15% de los pacientes con OI entre los diferentes fenotipos.
{{cite book}}: |work=ignorado ( ayuda ){{cite book}}: |website=ignorado ( ayuda ) Última actualización: 3 de febrero de 2019.En 1979 Sillence propuso una clasificación de la Osteogénesis Imperfecta (OI) en OI tipos I, II, III y IV.
En 2004 y 2007, esta clasificación se amplió con los tipos de OI V a VIII debido a características clínicas distintas y/o diferentes mutaciones genéticas causales.
Por ejemplo, puede ocurrir que a un feto se le diagnostique OI tipo II, la forma letal, en una ecografía prenatal, pero después del nacimiento resulte menos afectado de lo que se pensaba inicialmente y eventualmente se reclasifica como OI tipo III o incluso tipo VI, si se inicia un tratamiento adecuado desde el principio.
La esclerótica azul pálida persistente es poco común en la OI tipo IV, pero se puede observar hasta en el 10% de los sujetos afectados.
Los Hospitales Shriners para Niños se comprometen a brindar atención especializada a niños con afecciones ortopédicas [...] independientemente de la capacidad de pago de las familias.