Un prión / ˈ p r iː ɒ n / ⓘ es unaproteína mal plegadaque puede inducir el plegamiento incorrecto de variantes normales de la misma proteína y desencadenarla muerte celular. Los priones causan enfermedades priónicas conocidas comoencefalopatías espongiformes transmisiblesenfermedades neurodegenerativastransmisibles y fatalesen humanos y animales. [3][4]Las proteínas pueden plegarse mal esporádicamente, debido amutaciones genéticaso por exposición a una proteína que ya está mal plegada. [5]La consiguienteestructura tridimensionalles confiere la capacidad de provocar un plegamiento incorrecto de otras proteínas.
La palabra prión se deriva del término "partícula infecciosa proteica". [6] [7] El papel hipotético de una proteína como agente infeccioso contrasta con todos los demás agentes infecciosos conocidos, como viroides , virus , bacterias , hongos y parásitos , todos los cuales contienen ácidos nucleicos ( ADN , ARN o ambos).
La mayoría de los priones son isoformas retorcidas de la proteína priónica principal (PrP), una proteína natural cuya función normal es incierta. Se plantea la hipótesis de que sean la causa de las encefalopatías espongiformes transmisibles (EET), [8] incluida la tembladera en ovejas, la emaciación crónica (CWD) en ciervos, la encefalopatía espongiforme bovina (EEB) en bovinos (enfermedad de las vacas locas), la encefalopatía espongiforme felina (EEF) ) en felinos y la enfermedad de Creutzfeldt-Jakob (ECJ) en humanos.
Todas las enfermedades priónicas conocidas en los mamíferos afectan la estructura del cerebro u otro tejido neural ; todos son progresivos, no se conoce ningún tratamiento eficaz y siempre son mortales . [9] Todas las enfermedades priónicas de los mamíferos conocidas fueron causadas por PrP hasta 2015, cuando se planteó la hipótesis de que una forma priónica de alfa-sinucleína causaba atrofia sistémica múltiple (MSA). [10]
Los priones son un tipo de proteína intrínsecamente desordenada , que cambian continuamente su conformación a menos que estén unidos a una pareja específica como otra proteína. Con un prión, dos cadenas de proteínas se estabilizan si una se une a otra en la misma conformación. La probabilidad de que esto suceda es baja, pero una vez que sucede, la combinación de ambos es muy estable. Luego se pueden agregar más unidades, formando una especie de " fibrilla ". [11] Los priones forman agregados anormales de proteínas llamadas amiloides , que se acumulan en el tejido infectado y se asocian con daño tisular y muerte celular. [12] Los amiloides también están asociados con varias otras enfermedades neurodegenerativas, como la enfermedad de Alzheimer y la enfermedad de Parkinson . [13] [14]
Una enfermedad priónica es un tipo de proteopatía o enfermedad de proteínas estructuralmente anormales. En los seres humanos, se cree que los priones son la causa de la enfermedad de Creutzfeldt-Jakob (CJD), su variante (vCJD), el síndrome de Gerstmann-Sträussler-Scheinker (GSS), el insomnio familiar fatal (FFI) y el kuru . [15] También hay evidencia que sugiere que los priones pueden desempeñar un papel en el proceso de la enfermedad de Alzheimer, la enfermedad de Parkinson y la esclerosis lateral amiotrófica (ELA); éstas se han denominado enfermedades similares a priones . [16] [17] [18] [19] También se han identificado varias proteínas de levadura con propiedades prionógenas, [20] [21] así como una proteína implicada en la modificación de las sinapsis durante la formación de recuerdos [22] [11 ] (ver Eric Kandel § Cambios moleculares durante el aprendizaje ). La replicación de priones está sujeta a epimutación y selección natural al igual que otras formas de replicación, y su estructura varía ligeramente entre especies. [23]
Los agregados de priones son estables, y esta estabilidad estructural significa que los priones son resistentes a la desnaturalización por agentes químicos y físicos: no pueden destruirse mediante desinfección o cocción ordinaria. Esto dificulta la eliminación y contención de estas partículas, y el riesgo de propagación iatrogénica a través de instrumentos médicos es una preocupación creciente.
La palabra prión , acuñada en 1982 por Stanley B. Prusiner , se deriva de proteína e infección , de ahí prión , [24] [25] y es la abreviatura de "partícula proteica infecciosa", [10] en referencia a su capacidad para autopropagarse y transmitir su conformación a otras proteínas. [26] Su pronunciación principal es / ˈ p r iː ɒ n / ⓘ ,[27][28][29]aunque / ˈ p r aɪ ɒ n / , comose pronuncianombrehomográficoavetambién se escucha[29][30]En su artículo de 1982 introduciendo el término, Prusiner especificó que se "pronunciapree-on". [31]
La principal proteína priónica (PrP) de la que están hechos los priones se encuentra en todo el cuerpo, incluso en personas y animales sanos. Sin embargo, la PrP que se encuentra en el material infeccioso tiene una estructura diferente y es resistente a las proteasas , las enzimas del cuerpo que normalmente pueden descomponer las proteínas. La forma normal de la proteína se llama PrP C , mientras que la forma infecciosa se llama PrP Sc : la C se refiere a la PrP "celular", mientras que la Sc se refiere a la " tembladera ", la enfermedad priónica prototípica que se presenta en las ovejas. [32] Si bien la PrP C está estructuralmente bien definida, la PrP Sc es ciertamente polidispersa y está definida en un nivel relativamente pobre. Se puede inducir que la PrP se pliegue en otras isoformas más o menos bien definidas in vitro, y su relación con las formas patógenas in vivo aún no está clara. [ cita necesaria ]
La PrP C es una proteína normal que se encuentra en las membranas de las células , "incluidos varios componentes sanguíneos de los cuales las plaquetas constituyen el mayor reservorio en los humanos". [33] Tiene 209 aminoácidos (en humanos), un enlace disulfuro , una masa molecular de 35 a 36 kDa y una estructura principalmente alfa-helicoidal . Existen varias formas topológicas ; una forma de superficie celular anclada a través de glicolípidos y dos formas transmembrana . [34] La proteína normal no es sedimentable; lo que significa que no puede separarse mediante técnicas de centrifugación. [35] Su función es un tema complejo que continúa siendo investigado. PrP C se une a iones de cobre (II) con alta afinidad. [36] La importancia de este hallazgo no está clara, pero se supone que se relaciona con la estructura o función de la PrP. La PrP C es fácilmente digerida por la proteinasa K y puede liberarse de la superficie celular in vitro mediante la enzima fosfoinositido fosfolipasa C (PI-PLC), que escinde el anclaje glicolípido glicofosfatidilinositol (GPI). [37] Se ha informado que la PrP desempeña funciones importantes en la adhesión célula-célula y la señalización intracelular in vivo y, por lo tanto, puede estar involucrada en la comunicación célula-célula en el cerebro. [38]
La proteína similar a la PrP Sc resistente a proteasa (PrP res ) es el nombre que se le da a cualquier isoforma de PrP c que se altera estructuralmente y se convierte en una forma resistente a la proteinasa K mal plegada in vitro . [39] Para modelar la conversión de PrP C a PrP Sc in vitro , Saborio et al . convirtió rápidamente PrP C en PrP res mediante un procedimiento que implica la amplificación cíclica del plegamiento incorrecto de la proteína . [40] El término "PrP res " se utiliza para distinguir la PrP mal plegada creada in vitro de la PrP Sc , que se aísla de tejido infeccioso y se asocia con el agente de la encefalopatía espongiforme transmisible. [41] Por ejemplo, a diferencia de la PrP Sc , la PrP res puede no ser necesariamente infecciosa.

La isoforma infecciosa de PrP, conocida como PrP Sc , o simplemente prión, es capaz de convertir proteínas PrP C normales en la isoforma infecciosa cambiando su conformación o forma; esto, a su vez, altera la forma en que se interconectan las proteínas. PrP Sc siempre causa enfermedad priónica. Aunque se desconoce la estructura 3D exacta de PrP Sc , tiene una mayor proporción de estructura de lámina β en lugar de la estructura de hélice α normal . [42] Las agregaciones de estas isoformas anormales forman fibras amiloides altamente estructuradas , que se acumulan para formar placas. El extremo de cada fibra actúa como plantilla a la que se pueden unir moléculas de proteína libres, permitiendo que la fibra crezca. En la mayoría de las circunstancias, sólo se incorporan a la fibra en crecimiento moléculas de PrP con una secuencia de aminoácidos idéntica a la PrP Sc infecciosa. [35] Sin embargo, también es posible una rara transmisión entre especies. [43]
La función fisiológica de la proteína priónica sigue siendo poco conocida. Si bien los datos de experimentos in vitro sugieren muchas funciones diferentes, los estudios en ratones con desactivación de PrP han proporcionado sólo información limitada porque estos animales sólo presentan anomalías menores. En una investigación realizada en ratones, se descubrió que la escisión de PrP en los nervios periféricos provoca la activación de la reparación de la mielina en las células de Schwann y que la falta de proteínas PrP provocaba la desmielinización en esas células. [44]
MAVS, RIP1 y RIP3 son proteínas similares a priones que se encuentran en otras partes del cuerpo. También se polimerizan en fibras amiloides filamentosas que inician la muerte celular regulada en el caso de una infección viral para evitar la propagación de viriones a otras células circundantes. [45]
Una revisión de la evidencia realizada en 2005 sugirió que la PrP puede tener una función normal en el mantenimiento de la memoria a largo plazo . [46] Además, un estudio de 2004 encontró que los ratones que carecen de genes para la proteína PrP celular normal muestran una potenciación alterada del hipocampo a largo plazo . [47] [48] Un estudio reciente que también sugiere por qué este podría ser el caso, encontró que la proteína neuronal CPEB tiene una secuencia genética similar a las proteínas priónicas de levadura. La formación priónica de CPEB es esencial para mantener los cambios sinápticos a largo plazo asociados con la formación de la memoria a largo plazo. [49]
Un artículo de 2006 del Instituto Whitehead de Investigación Biomédica indica que la expresión de PrP en células madre es necesaria para la autorrenovación de la médula ósea de un organismo . El estudio demostró que todas las células madre hematopoyéticas a largo plazo expresan PrP en su membrana celular y que los tejidos hematopoyéticos con células madre sin PrP exhiben una mayor sensibilidad al agotamiento celular. [50]
Existe cierta evidencia de que la PrP puede desempeñar un papel en la inmunidad innata , ya que la expresión de PRNP , el gen PrP, está regulada positivamente en muchas infecciones virales y la PrP tiene propiedades antivirales contra muchos virus, incluido el VIH . [51]

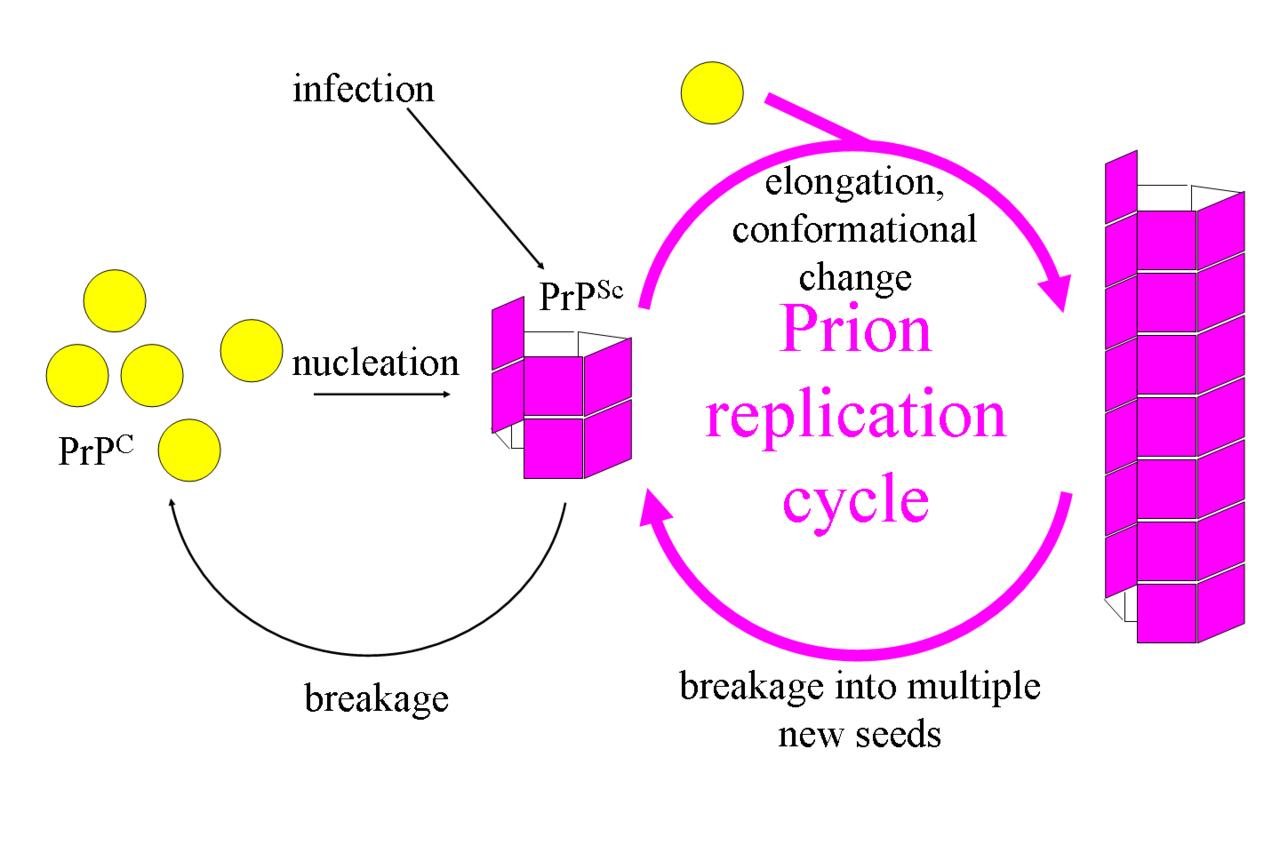
La primera hipótesis que intentó explicar cómo los priones se replican únicamente a partir de proteínas fue el modelo de heterodímero . [52] Este modelo asumió que una sola molécula de PrP Sc se une a una sola molécula de PrP C y cataliza su conversión en PrP Sc . Las dos moléculas de PrP Sc luego se separan y pueden convertir más PrP C. Sin embargo, un modelo de replicación de priones debe explicar cómo se propagan los priones y por qué su aparición espontánea es tan rara. Manfred Eigen demostró que el modelo de heterodímero requiere que PrPSc sea un catalizador extraordinariamente eficaz, aumentando la velocidad de la reacción de conversión en un factor de alrededor de 10 15 . [53] Este problema no surge si la PrP Sc existe solo en formas agregadas como el amiloide , donde la cooperatividad puede actuar como una barrera para la conversión espontánea. Es más, a pesar de un esfuerzo considerable, nunca se ha aislado la PrP Sc monomérica infecciosa. [ cita necesaria ]
Un modelo alternativo supone que la PrP Sc existe sólo como fibrillas y que los extremos de las fibrillas se unen a la PrP C y la convierten en PrP Sc . Si esto fuera todo, entonces la cantidad de priones aumentaría linealmente , formando fibrillas cada vez más largas. Pero durante la enfermedad priónica se observa un crecimiento exponencial tanto de PrP Sc como de la cantidad de partículas infecciosas . [54] [55] [56] Esto se puede explicar teniendo en cuenta la rotura de fibrillas. [57] Se ha encontrado una solución matemática para la tasa de crecimiento exponencial resultante de la combinación de crecimiento y rotura de fibrillas. [58] La tasa de crecimiento exponencial depende en gran medida de la raíz cuadrada de la concentración de PrP C. [58] El período de incubación está determinado por la tasa de crecimiento exponencial, y los datos in vivo sobre enfermedades priónicas en ratones transgénicos coinciden con esta predicción. [58] La misma dependencia de la raíz cuadrada también se observa in vitro en experimentos con una variedad de proteínas amiloides diferentes . [59]
El mecanismo de replicación de priones tiene implicaciones para el diseño de fármacos. Dado que el período de incubación de las enfermedades priónicas es tan largo, un fármaco eficaz no necesita eliminar todos los priones, sino simplemente reducir la velocidad de crecimiento exponencial. Los modelos predicen que la forma más eficaz de lograrlo, utilizando un fármaco con la dosis más baja posible, es encontrar un fármaco que se una a los extremos de las fibrillas e impida que sigan creciendo. [60]
Investigadores del Dartmouth College descubrieron que las moléculas cofactor endógenas del huésped, como la molécula de fosfolípidos (p. ej., fosfatidiletanolamina) y los polianiones (p. ej., moléculas de ARN monocatenario), son necesarias para formar moléculas de PrP Sc con altos niveles de infectividad específica in vitro , mientras que la PrP Sc de proteína sola Las moléculas parecen carecer de niveles significativos de infectividad biológica. [61] [62]
Los priones causan enfermedades neurodegenerativas al agregarse extracelularmente dentro del sistema nervioso central para formar placas conocidas como amiloides , que alteran la estructura normal del tejido . Esta alteración se caracteriza por "agujeros" en el tejido con la arquitectura esponjosa resultante debido a la formación de vacuolas en las neuronas. [68] Otros cambios histológicos incluyen astrogliosis y la ausencia de una reacción inflamatoria . [69] Si bien el período de incubación de las enfermedades priónicas es relativamente largo (de 5 a 20 años), una vez que aparecen los síntomas, la enfermedad progresa rápidamente y provoca daño cerebral y muerte. [70] Los síntomas neurodegenerativos pueden incluir convulsiones , demencia , ataxia (disfunción del equilibrio y la coordinación) y cambios de comportamiento o personalidad. [ cita necesaria ]
Muchas especies diferentes de mamíferos pueden verse afectadas por enfermedades priónicas, ya que la proteína priónica (PrP) es muy similar en todos los mamíferos. [71] Debido a las pequeñas diferencias en PrP entre diferentes especies, es inusual que una enfermedad priónica se transmita de una especie a otra. Sin embargo, se cree que la variante de la enfermedad priónica humana, la enfermedad de Creutzfeldt-Jakob, es causada por un prión que típicamente infecta al ganado, causando encefalopatía espongiforme bovina y se transmite a través de la carne infectada. [72]
Todas las enfermedades priónicas conocidas son intratables y mortales. [73] Sin embargo, una vacuna desarrollada en ratones puede proporcionar información sobre cómo proporcionar una vacuna para resistir las infecciones por priones en humanos. [74] Además, en 2006, los científicos anunciaron que habían modificado genéticamente ganado que carecía de un gen necesario para la producción de priones, lo que en teoría los hacía inmunes a la EEB, [75] basándose en investigaciones que indican que los ratones que carecen de la proteína priónica normal son resistentes a la infección por proteína priónica de la tembladera. [76] En 2013, un estudio reveló que 1 de cada 2000 personas en el Reino Unido podría albergar la proteína priónica infecciosa que causa la vCJD. [77]
Hasta 2015, se consideraba que todas las enfermedades priónicas de los mamíferos conocidas estaban causadas por la proteína priónica, PrP ; en 2015 se descubrió que la atrofia multisistémica era transmisible y se planteó la hipótesis de que era causada por un nuevo prión, la forma mal plegada de una proteína llamada alfa-sinucleína . [10] La forma endógena y correctamente plegada de la proteína priónica se denomina PrP C (por común o celular ) , mientras que la forma mal plegada y vinculada a una enfermedad se denomina PrP Sc (por Sc rapie ), después de una de las enfermedades primero . vinculado a priones y neurodegeneración. [35] [16] Se desconoce la estructura precisa del prión, aunque se pueden formar espontáneamente combinando PrP C , ácido poliadenílico homopolimérico y lípidos en una reacción de amplificación cíclica de plegamiento incorrecto de proteínas (PMCA), incluso en ausencia de pre- priones infecciosos existentes. [61] Este resultado es una prueba más de que la replicación de priones no requiere información genética. [78]
Se ha reconocido que las enfermedades priónicas pueden surgir de tres formas diferentes: adquirida, familiar o esporádica. [79] A menudo se supone que la forma enferma interactúa directamente con la forma normal para hacer que reorganice su estructura. Una idea, la hipótesis de la "Proteína X", es que una proteína celular aún no identificada (Proteína X) permite la conversión de PrP C en PrP Sc al reunir una molécula de cada una de las dos en un complejo. [80]
El principal método de infección en animales es mediante la ingestión. Se cree que los priones pueden depositarse en el medio ambiente a través de los restos de animales muertos y a través de la orina, la saliva y otros fluidos corporales. Luego pueden permanecer en el suelo uniéndose a la arcilla y otros minerales. [81]
Un equipo de investigación de la Universidad de California ha proporcionado evidencia que respalda la teoría de que la infección puede ocurrir a partir de priones en el estiércol. [82] Y, dado que el estiércol está presente en muchas áreas que rodean los depósitos de agua, así como también se utiliza en muchos campos de cultivo, aumenta la posibilidad de una transmisión generalizada. En enero de 2011 se informó que los investigadores habían descubierto priones que se propagaban a través de la transmisión aérea en partículas de aerosol , en un experimento de pruebas con animales centrado en la infección por scrapie en ratones de laboratorio . [83] En 2011 se publicó evidencia preliminar que respalda la idea de que los priones pueden transmitirse mediante el uso de gonadotropina menopáusica humana derivada de la orina , administrada para el tratamiento de la infertilidad . [84]
En 2015, investigadores del Centro de Ciencias de la Salud de la Universidad de Texas en Houston descubrieron que las plantas pueden ser vectores de priones. Cuando los investigadores alimentaron a los hámsteres con pasto que crecía en el suelo donde estaba enterrado un ciervo que murió con caquexia crónica (CWD), los hámsteres enfermaron con caquexia crónica, lo que sugiere que los priones pueden unirse a las plantas, que luego los llevan a la estructura de las hojas y el tallo. , donde pueden ser comidos por los herbívoros, completando así el ciclo. Por tanto, es posible que se vaya acumulando progresivamente un número de priones en el medio ambiente. [85] [86]
Las partículas infecciosas que poseen ácido nucleico dependen de él para dirigir su replicación continua. Los priones, sin embargo, son infecciosos por su efecto sobre las versiones normales de la proteína. Por lo tanto, la esterilización de priones requiere la desnaturalización de la proteína a un estado en el que la molécula ya no sea capaz de inducir el plegamiento anormal de las proteínas normales. En general, los priones son bastante resistentes a las proteasas , el calor, la radiación ionizante y los tratamientos con formaldehído , [87] aunque su infectividad puede reducirse con dichos tratamientos. La descontaminación eficaz de priones se basa en la hidrólisis de las proteínas o en la reducción o destrucción de la estructura terciaria de las proteínas . Los ejemplos incluyen hipoclorito de sodio , hidróxido de sodio y detergentes fuertemente ácidos como el LpH. [88]
La Organización Mundial de la Salud recomienda cualquiera de los tres procedimientos siguientes para la esterilización de todos los instrumentos quirúrgicos resistentes al calor para garantizar que no estén contaminados con priones:
Se ha descubierto que 134 °C (273 °F) durante 18 minutos en un autoclave de vapor presurizado es algo eficaz para desactivar el agente patógeno. [90] [91] La esterilización con ozono se está estudiando actualmente como un método potencial para la desnaturalización y desactivación de priones. [92] Otros enfoques que se están desarrollando incluyen tiourea - tratamiento con urea , tratamiento con cloruro de guanidinio , [93] y subtilisina especial resistente al calor combinada con calor y detergente. [94] Un método suficiente para esterilizar priones en un material puede fallar en otro. [95]
Aún no se ha logrado la renaturalización de un prión completamente desnaturalizado a un estado infeccioso; sin embargo, los priones parcialmente desnaturalizados pueden renaturalizarse hasta alcanzar un estado infeccioso en determinadas condiciones artificiales. [96]
Una evidencia abrumadora muestra que los priones resisten la degradación y persisten en el medio ambiente durante años, y las proteasas no los degradan. La evidencia experimental muestra que los priones libres se degradan con el tiempo, mientras que los priones adheridos al suelo permanecen en niveles estables o crecientes, lo que sugiere que es probable que los priones se acumulen en el medio ambiente. [97] [98] Un estudio de 2015 realizado por científicos estadounidenses encontró que el secado y la humectación repetidos pueden hacer que los priones adheridos al suelo sean menos infecciosos, aunque esto dependía del tipo de suelo al que estaban adheridos. [99]
En algunos hongos también se encuentran proteínas que muestran un comportamiento de tipo priónico , lo que ha resultado útil para ayudar a comprender los priones de los mamíferos. Los priones de los hongos no siempre causan enfermedades en sus huéspedes. [100] En la levadura, el replegamiento de proteínas a la configuración priónica es ayudado por proteínas chaperonas como Hsp104 . [21] Todos los priones conocidos inducen la formación de un pliegue amiloide , en el que la proteína se polimeriza en un agregado que consiste en láminas beta muy compactas . Los agregados de amiloide son fibrillas que crecen en sus extremos y se replican cuando la rotura hace que dos extremos en crecimiento se conviertan en cuatro extremos en crecimiento. El período de incubación de las enfermedades priónicas está determinado por la tasa de crecimiento exponencial asociada con la replicación de priones, que es un equilibrio entre el crecimiento lineal y la rotura de agregados. [58]
A principios de los años 90, Reed Wickner descubrió en la levadura Saccharomyces cerevisiae proteínas fúngicas que exhibían cambios conformacionales tipo plantilla [ se necesita más explicación ] . Por su similitud mecánica con los priones de mamíferos, se les denominó priones de levadura . Posteriormente, también se encontró un prión en el hongo Podospora anserina . Estos priones se comportan de manera similar a la PrP, pero, en general, no son tóxicos para sus huéspedes. El grupo de Susan Lindquist en el Instituto Whitehead ha argumentado que algunos de los priones fúngicos no están asociados con ningún estado patológico, pero pueden tener un papel útil; sin embargo, los investigadores del NIH también han aportado argumentos que sugieren que los priones de los hongos podrían considerarse un estado de enfermedad. [101] Existe evidencia de que las proteínas fúngicas han desarrollado funciones específicas que son beneficiosas para el microorganismo y que mejoran su capacidad para adaptarse a sus diversos entornos. [102] Además, dentro de las levaduras, los priones pueden actuar como vectores de herencia epigenética , transfiriendo rasgos a la descendencia sin ningún cambio genómico . [103] [104]
La investigación sobre priones fúngicos ha brindado un fuerte apoyo al concepto de proteína exclusiva, ya que se ha demostrado que la proteína purificada extraída de células con un estado priónico convierte la forma normal de la proteína en una forma mal plegada in vitro y, en el proceso, preserva la información correspondiente a diferentes cepas del estado priónico. También ha arrojado algo de luz sobre los dominios priónicos, que son regiones de una proteína que promueven la conversión en prión. Los priones fúngicos han ayudado a sugerir mecanismos de conversión que pueden aplicarse a todos los priones, aunque los priones fúngicos parecen distintos de los priones infecciosos de mamíferos por la falta del cofactor necesario para la propagación. Los dominios priónicos característicos pueden variar entre especies; por ejemplo, los dominios priónicos característicos de los hongos no se encuentran en los priones de los mamíferos. [ cita necesaria ]
No existen tratamientos eficaces para las enfermedades priónicas. [106] Los ensayos clínicos en humanos no han tenido éxito y se han visto obstaculizados por la rareza de las enfermedades priónicas. [106] Aunque algunos tratamientos potenciales se han mostrado prometedores en el laboratorio, ninguno ha sido efectivo una vez que la enfermedad ha comenzado. [107]
Se han encontrado dominios similares a priones en una variedad de otras proteínas de mamíferos. Algunas de estas proteínas han sido implicadas en la ontogenia de trastornos neurodegenerativos relacionados con la edad, como la esclerosis lateral amiotrófica (ELA), la degeneración del lóbulo frontotemporal con inclusiones positivas de ubiquitina (FTLD-U), la enfermedad de Alzheimer , la enfermedad de Parkinson y la enfermedad de Huntington . [108] [18] [17] También están implicados en algunas formas de amiloidosis sistémica , incluida la amiloidosis AA , que se desarrolla en humanos y animales con enfermedades inflamatorias e infecciosas como tuberculosis , enfermedad de Crohn , artritis reumatoide y VIH/SIDA . La amiloidosis AA, al igual que la enfermedad priónica, puede ser transmisible. [109] Esto ha dado lugar al "paradigma de los priones", en el que proteínas que de otro modo serían inofensivas pueden convertirse en una forma patógena mediante un pequeño número de proteínas nucleadas mal plegadas. [110]
La definición de dominio similar a un prión surge del estudio de los priones de hongos. En la levadura, las proteínas prionógenas tienen un dominio priónico portátil que es necesario y suficiente para la autoformación de plantillas y la agregación de proteínas. Esto se ha demostrado uniendo el dominio priónico a una proteína informadora, que luego se agrega como un prión conocido. De manera similar, la eliminación del dominio priónico de una proteína priónica fúngica inhibe la prionogénesis. Esta visión modular del comportamiento de los priones ha llevado a la hipótesis de que dominios priónicos similares están presentes en las proteínas animales, además de la PrP. [108] Estos dominios priónicos fúngicos tienen varios rasgos de secuencia característicos. Por lo general, están enriquecidos en residuos de asparagina, glutamina, tirosina y glicina, siendo particularmente propicio el sesgo de asparagina a la propiedad agregativa de los priones. Históricamente, se ha considerado que la prionogénesis es independiente de la secuencia y sólo depende del contenido relativo de residuos. Sin embargo, se ha demostrado que esto es falso, ya que se ha demostrado que el espaciamiento de prolinas y residuos cargados es crítico en la formación de amiloide. [20]
Los análisis bioinformáticos han predicho que más de 250 proteínas humanas contienen dominios similares a priones (PrLD). Se supone que estos dominios tienen las mismas propiedades amiloidogénicas transmisibles que la PrP y las proteínas fúngicas conocidas. Al igual que en la levadura, las proteínas implicadas en la expresión genética y la unión del ARN parecen estar particularmente enriquecidas en las PrLD, en comparación con otras clases de proteínas. En particular, 29 de las 210 proteínas conocidas con un motivo de reconocimiento de ARN también tienen un supuesto dominio priónico. Mientras tanto, varias de estas proteínas de unión a ARN han sido identificadas de forma independiente como patógenas en casos de ELA, FTLD-U, enfermedad de Alzheimer y enfermedad de Huntington. [111]
Se plantea la hipótesis de que la patogenicidad de los priones y las proteínas con dominios similares a priones surge de su capacidad de automodelación y del resultante crecimiento exponencial de las fibrillas de amiloide. La presencia de fibrillas de amiloide en pacientes con enfermedades degenerativas ha sido bien documentada. Estas fibrillas de amiloide se consideran el resultado de proteínas patógenas que se autopropagan y forman agregados no funcionales altamente estables. [111] Si bien esto no implica necesariamente una relación causal entre las enfermedades amiloides y degenerativas, la toxicidad de ciertas formas de amiloide y la sobreproducción de amiloide en casos familiares de trastornos degenerativos apoya la idea de que la formación de amiloide es generalmente tóxica. [112]
Específicamente, se ha encontrado agregación de TDP-43 , una proteína de unión a ARN, en pacientes con ELA/EMN, y se han identificado mutaciones en los genes que codifican estas proteínas en casos familiares de ELA/EMN. Estas mutaciones promueven el plegamiento incorrecto de las proteínas en una conformación similar a un prión. La forma mal plegada de TDP-43 forma inclusiones citoplasmáticas en las neuronas afectadas y se encuentra agotada en el núcleo. Además de ALS/MND y FTLD-U, la patología TDP-43 es una característica de muchos casos de enfermedad de Alzheimer, enfermedad de Parkinson y enfermedad de Huntington. El mal plegamiento de TDP-43 está dirigido en gran medida por su dominio priónico. Este dominio es inherentemente propenso a plegarse incorrectamente, mientras que se ha descubierto que las mutaciones patológicas en TDP-43 aumentan esta propensión a plegarse incorrectamente, lo que explica la presencia de estas mutaciones en casos familiares de ELA/EMN. Al igual que en la levadura, se ha demostrado que el dominio priónico de TDP-43 es necesario y suficiente para el plegamiento incorrecto y la agregación de proteínas. [108]
De manera similar, se han identificado mutaciones patógenas en los dominios priónicos de las riboproteínas nucleares heterogéneas hnRNPA2B1 y hnRNPA1 en casos familiares de degeneración de músculos, cerebro, huesos y neuronas motoras. La forma natural de todas estas proteínas muestra una tendencia a autoensamblarse en fibrillas de amiloide, mientras que las mutaciones patógenas exacerban este comportamiento y conducen a una acumulación excesiva. [113]
En teoría, los priones podrían emplearse como agentes armados . [114] [115] Con tasas de mortalidad potenciales del 100%, los priones podrían ser un arma biológica eficaz, a veces llamada "arma bioquímica", porque un prión es un bioquímico. Un aspecto desfavorable son los períodos de incubación muy largos de los priones. La exposición intensa y persistente de priones al intestino podría acortar el inicio general. [116] Otro aspecto del uso de priones en la guerra es la dificultad de detección y descontaminación . [117]
En los siglos XVIII y XIX, se observó que la exportación de ovejas desde España coincidía con una enfermedad llamada scrapie . Esta enfermedad provocaba que los animales afectados "se tumbaran, se mordieran las patas y los pies, se frotaran el lomo contra los postes, no medraran, dejaran de alimentarse y finalmente quedaran cojos" . [118] También se observó que la enfermedad tiene un largo período de incubación que es una característica clave de las encefalopatías espongiformes transmisibles (EET) . Aunque en aquel entonces se desconocía la causa de la tembladera, probablemente se trate de la primera encefalopatía espongiforme transmisible registrada. [ cita necesaria ]
En la década de 1950, Carleton Gajdusek inició una investigación que finalmente demostró que el kuru podía transmitirse a los chimpancés a través de lo que posiblemente era un nuevo agente infeccioso, trabajo por el que finalmente ganó el Premio Nobel en 1976 . Durante los años 1960, dos investigadores radicados en Londres, el radiobiólogo Tikvah Alper y el biofísico John Stanley Griffith , desarrollaron la hipótesis de que las encefalopatías espongiformes transmisibles son causadas por un agente infeccioso compuesto únicamente de proteínas. [119] [120] Investigaciones anteriores realizadas por EJ Field sobre scrapie y kuru habían encontrado evidencia de la transferencia de polisacáridos patológicamente inertes que solo se vuelven infecciosos después de la transferencia, en el nuevo huésped. [121] [122] Alper y Griffith querían explicar el descubrimiento de que el misterioso agente infeccioso que causa las enfermedades de la tembladera y la enfermedad de Creutzfeldt-Jakob resistía la radiación ionizante . [123] Griffith propuso tres formas en las que una proteína podría ser un patógeno . [124]
En la primera hipótesis , sugirió que si la proteína es el producto de un gen normalmente suprimido , y la introducción de la proteína podría inducir la expresión del gen, es decir, despertar el gen inactivo, entonces el resultado sería un proceso indistinguible de la replicación. ya que la expresión del gen produciría la proteína, que luego despertaría el gen en otras células . [ cita necesaria ]
Su segunda hipótesis forma la base de la teoría moderna de los priones y propuso que una forma anormal de una proteína celular puede convertir proteínas normales del mismo tipo en su forma anormal, lo que lleva a la replicación. [ cita necesaria ]
Su tercera hipótesis proponía que el agente podría ser un anticuerpo si el anticuerpo fuera su propio antígeno objetivo , ya que dicho anticuerpo daría como resultado que se produjeran cada vez más anticuerpos contra sí mismo. Sin embargo, Griffith reconoció que era poco probable que esta tercera hipótesis fuera cierta debido a la falta de una respuesta inmune detectable . [125]
Francis Crick reconoció la importancia potencial de la hipótesis de Griffith de la proteína exclusiva para la propagación de la tembladera en la segunda edición de su " Dogma central de biología molecular " (1970): Si bien afirmaba que el flujo de información de secuencia de proteína a proteína, o de proteína a El ARN y el ADN estaban "excluidos", señaló que la hipótesis de Griffith era una contradicción potencial (aunque Griffith no la promovió tanto). [126] La hipótesis revisada se formuló posteriormente, en parte, para dar cabida a la transcripción inversa (que tanto Howard Temin como David Baltimore descubrieron en 1970). [127]
En 1982, Stanley B. Prusiner de la Universidad de California, San Francisco , anunció que su equipo había purificado la hipotética proteína infecciosa, que no parecía estar presente en huéspedes sanos, aunque no consiguieron aislar la proteína hasta dos años. después del anuncio de Prusiner. [128] [31] La proteína recibió el nombre de prión, que significa "partícula infecciosa proteica", derivada de las palabras proteína e infección . Cuando se descubrió el prión, muchos apoyaron la primera hipótesis de Griffith de que la proteína era el producto de un gen normalmente silencioso. Sin embargo, posteriormente se descubrió que la misma proteína existe en los huéspedes normales pero en forma diferente. [129]
Tras el descubrimiento de la misma proteína en diferentes formas en individuos no infectados, la proteína específica de la que estaba compuesto el prión se denominó proteína priónica (PrP), y la segunda hipótesis de Griffith de que una forma anormal de una proteína del huésped puede convertir otras proteínas del mismo tipo en su forma anormal, se convirtió en la teoría dominante. [125] Prusiner recibió el Premio Nobel de Fisiología o Medicina en 1997 por su investigación sobre priones. [130] [131]
Los científicos dijeron ayer que han utilizado técnicas de ingeniería genética para producir el primer ganado que puede ser biológicamente incapaz de contraer la enfermedad de las vacas locas.
El Premio Nobel de Fisiología o Medicina de 1997 fue concedido a Stanley B. Prusiner "por su descubrimiento de los priones, un nuevo principio biológico de infección".
{kind=link}