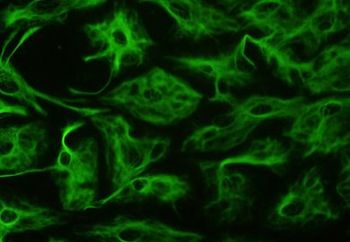
La enfermedad progresa hasta un desenlace mortal en la mayor parte de los casos.La enfermedad recibe el nombre por el patólogo neozelandés William Stuart Alexander, quien describió el síndrome en 1949 junto con la profesora Dorothy Rusell en el London Hospital.En principio, se observó la acumulación en el cerebro en torno a los astrocitos y a la barrera hematoencefálica de unas estructuras características eosinófilas y vermiformes que se llamaron fibras de Rosenthal, en honor al patólogo alemán Werner Rosenthal quien las describió en 1989 en la siringomielia.Estas estructuras eran ricas en PAFG y en una proteína de shock término llamada α-B-Cristalina.[10] Estos últimos se caracterizan porque en su citoesqueleto contienen un filamento intermedio, la ya mencionada PAFG, que al mutar construye una estructura proteica defectuosa.Las imágenes por microscopía electrónica muestran un vínculo estrecho entre las fibras de Rosenthal y los filamentos intermedios.Además existe desmielinización histológicamente hablando en los afectados tardíos, o ausencia de mielinización en los niños.
Localización de las principales mutaciones en la
PAFG
. De ellas, la P47L podría no estar asociada a la enfermedad. Los rectángulos representan los dominios α-hélice de la proteína.