Un prión / ˈ p r iː ɒ n /ⓘ es unaproteína mal plegadaque induce un plegamiento incorrecto en variantes normales de la misma proteína, lo que lleva ala muerte celular. Los priones son responsables de enfermedades priónicas, conocidas comoencefalopatías espongiformes transmisiblesenfermedades neurodegenerativasfatales y transmisiblesque afectan tanto a humanos como a animales.[3][4]Estas proteínas pueden plegarse incorrectamente esporádicamente, debido a mutaciones genéticas, o por exposición a una proteína ya mal plegada, lo que lleva a unaestructura tridimensionalque puede propagar el plegamiento incorrecto en otras proteínas.[5]
Todas las enfermedades priónicas conocidas en mamíferos afectan la estructura del cerebro u otros tejidos neuronales . Estas enfermedades son progresivas, no tienen un tratamiento efectivo conocido y son invariablemente fatales. [9] Se pensaba que la mayoría de las enfermedades priónicas eran causadas por PrP hasta 2015, cuando una forma priónica de alfa-sinucleína se relacionó con la atrofia multisistémica (MSA). [10] Los priones también están relacionados con otras enfermedades neurodegenerativas como la enfermedad de Alzheimer , la enfermedad de Parkinson y la esclerosis lateral amiotrófica (ELA), que a veces se denominan enfermedades similares a priones . [11] [12]
Los priones son un tipo de proteína intrínsecamente desordenada que cambia continuamente de conformación a menos que se una a un compañero específico, como otra proteína. Una vez que un prión se une a otro en la misma conformación, se estabiliza y puede formar una fibrilla , lo que da lugar a agregados proteicos anormales llamados amiloides . Estos amiloides se acumulan en el tejido infectado, lo que provoca daño y muerte celular. [13] La estabilidad estructural de los priones los hace resistentes a la desnaturalización por agentes químicos o físicos, lo que complica la eliminación y la contención, y genera preocupaciones sobre la propagación iatrogénica a través de instrumentos médicos.
Etimología y pronunciación
La palabra prión , acuñada en 1982 por Stanley B. Prusiner , se deriva de proteína e infección , de ahí prión , [14] y es la abreviatura de "partícula infecciosa proteínica", [10] en referencia a su capacidad de autopropagarse y transmitir su conformación a otras proteínas. [15] Su pronunciación principal es / ˈpr iːɒn /ⓘ ,[16][17][18]aunque/ ˈ p r aɪ ɒ n /se pronunciaelnombrehomográficoave[18]también se escucha.[19]En su artículo de 1982 en el que introdujo el término, Prusiner especificó que se "pronunciapree-on".[14]
Proteína priónica
Estructura
Los priones consisten en una forma mal plegada de la proteína priónica mayor (PrP), una proteína que es una parte natural de los cuerpos de los humanos y otros animales. La PrP que se encuentra en los priones infecciosos tiene una estructura diferente y es resistente a las proteasas , las enzimas del cuerpo que normalmente pueden descomponer las proteínas. La forma normal de la proteína se llama PrP C , mientras que la forma infecciosa se llama PrP Sc : la C se refiere a la PrP "celular", mientras que la Sc se refiere a la " scrapie ", la enfermedad priónica prototípica, que se produce en las ovejas. [20] La PrP también puede ser inducida a plegarse en otras isoformas más o menos bien definidas in vitro; aunque sus relaciones con la(s) forma(s) que son patógenas in vivo a menudo no están claras, los análisis estructurales de alta resolución han comenzado a revelar características estructurales que se correlacionan con la infectividad de los priones. [21]
PrP Sc (teñida en rojo) revelada en una fotomicrografía de células neuronales de ratón infectadas con scrapie.
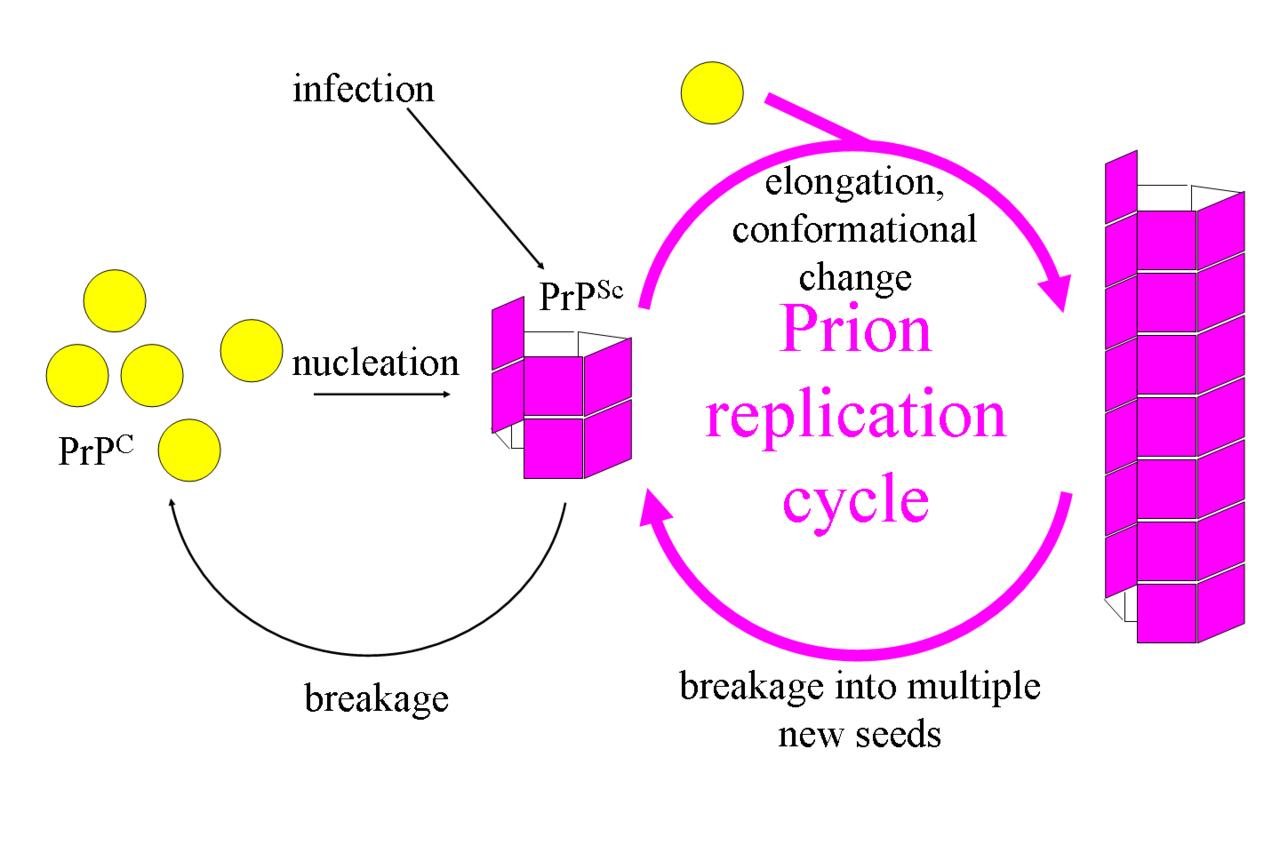
La isoforma infecciosa de PrP, conocida como PrP Sc , o simplemente el prión, es capaz de convertir las proteínas PrP C normales en la isoforma infecciosa al cambiar su conformación o forma; esto, a su vez, altera la forma en que las proteínas se interconectan . PrP Sc siempre causa enfermedad priónica. PrP Sc tiene una mayor proporción de estructura de lámina β en lugar de la estructura normal de hélice α . [31] [32] [33] Se han descubierto varias estructuras de PrP Sc altamente infecciosas derivadas del cerebro mediante criomicroscopía electrónica . [34] [35] [36] También se ha determinado otra estructura de fibrilla derivada del cerebro aislada de humanos con síndrome de Gerstmann-Straussler-Schienker . [37] Todas las estructuras descritas en alta resolución hasta ahora son fibras amiloides en las que las moléculas de PrP individuales se apilan a través de láminas beta intermoleculares. Sin embargo, también se han descrito matrices cristalinas 2-D con una resolución más baja en preparaciones ex vivo de priones. [38] En los amiloides priónicos, los anclajes de glucolípidos y los glicanos unidos a asparagina , cuando están presentes, se proyectan hacia afuera desde las superficies laterales de los núcleos de las fibras. A menudo, la PrP Sc está unida a las membranas celulares, presumiblemente a través de su matriz de anclajes de glucolípidos, sin embargo, a veces las fibras se disocian de las membranas y se acumulan fuera de las células en forma de placas. El extremo de cada fibra actúa como una plantilla sobre la que se pueden unir moléculas de proteína libre, lo que permite que la fibra crezca. Este proceso de crecimiento requiere un replegamiento completo de PrP C . [39] Diferentes cepas de priones tienen plantillas o conformaciones distintas, incluso cuando están compuestas de moléculas de PrP de la misma secuencia de aminoácidos , como ocurre en un genotipo de huésped particular . [40] [41] [42] [43] [44] En la mayoría de las circunstancias, solo las moléculas de PrP con una secuencia de aminoácidos idéntica a la PrP Sc infecciosa se incorporan a la fibra en crecimiento. [26] Sin embargo, la transmisión entre especies también ocurre raramente. [45]
PrPres
La proteína similar a PrP Sc resistente a la proteasa (PrP res ) es el nombre que se le da a cualquier isoforma de PrP c que se altera estructuralmente y se convierte en una forma resistente a la proteinasa K mal plegada . [46] Para modelar la conversión de PrP C a PrP Sc in vitro , Kocisko et al . demostraron que PrP Sc podría hacer que PrP C se convierta en PrP res en condiciones libres de células [47] y Soto et al . demostraron una amplificación sostenida de PrP res y la infectividad priónica mediante un procedimiento que implica la amplificación cíclica del mal plegamiento de proteínas . [48] El término "PrP res " puede referirse a formas resistentes a la proteasa de PrP Sc , que se aísla del tejido infeccioso y se asocia con el agente de la encefalopatía espongiforme transmisible, o a otras formas resistentes a la proteasa de PrP que, por ejemplo, podrían generarse in vitro . [49] En consecuencia, a diferencia de PrP Sc , PrP res puede no ser necesariamente infecciosa.
Modelos de formas normales (PrP C ) e infecciosas (PrP Sc ) de proteína priónica en una membrana: polipéptido (turquesa); glicanos (rojo); anclajes de glucolípidos (azul). Las estructuras centrales se basan en espectroscopia de RMN (PrP C ) y microscopía crioelectrónica (PrP Sc ).
Función normal de PrP
La función fisiológica de la proteína priónica sigue siendo poco conocida. Si bien los datos de experimentos in vitro sugieren muchas funciones diferentes, los estudios en ratones deficientes en PrP han proporcionado solo información limitada porque estos animales solo presentan anomalías menores. En investigaciones realizadas en ratones, se descubrió que la escisión de PrP en los nervios periféricos provoca la activación de la reparación de la mielina en las células de Schwann y que la falta de proteínas PrP causaba la desmielinización en esas células. [50]
PrP y muerte celular regulada
MAVS, RIP1 y RIP3 son proteínas similares a priones que se encuentran en otras partes del cuerpo. También se polimerizan en fibras amiloides filamentosas que inician la muerte celular regulada en caso de una infección viral para evitar la propagación de viriones a otras células circundantes. [51]
PrP y memoria a largo plazo
Una revisión de la evidencia en 2005 sugirió que la PrP puede tener una función normal en el mantenimiento de la memoria a largo plazo . [52] Además, un estudio de 2004 encontró que los ratones que carecen de genes para la proteína PrP celular normal muestran una potenciación a largo plazo alterada en el hipocampo . [53] [54] Un estudio reciente que también sugiere por qué este podría ser el caso, encontró que la proteína neuronal CPEB tiene una secuencia genética similar a las proteínas priónicas de levadura. La formación similar a la priónica de CPEB es esencial para mantener los cambios sinápticos a largo plazo asociados con la formación de la memoria a largo plazo. [55]
PrP y renovación de células madre
Un artículo de 2006 del Instituto Whitehead de Investigación Biomédica indica que la expresión de PrP en las células madre es necesaria para la autorrenovación de la médula ósea de un organismo. El estudio mostró que todas las células madre hematopoyéticas de largo plazo expresan PrP en su membrana celular y que los tejidos hematopoyéticos con células madre sin PrP muestran una mayor sensibilidad al agotamiento celular. [56]
PrP y la inmunidad innata
Hay algunas evidencias de que la PrP puede desempeñar un papel en la inmunidad innata , ya que la expresión de PRNP , el gen PrP, está regulada positivamente en muchas infecciones virales y la PrP tiene propiedades antivirales contra muchos virus, incluido el VIH . [57]
Replicación
Modelo heterodímero de propagación de prionesModelo fibrilar de propagación de priones.
La primera hipótesis que intentó explicar cómo los priones se replican de una manera solo proteica fue el modelo heterodímero . [58] Este modelo asumió que una sola molécula de PrP Sc se une a una sola molécula de PrP C y cataliza su conversión en PrP Sc . Las dos moléculas de PrP Sc luego se separan y pueden continuar convirtiendo más PrP C. Sin embargo, un modelo de replicación de priones debe explicar tanto cómo se propagan los priones como por qué su aparición espontánea es tan rara. Manfred Eigen demostró que el modelo heterodímero requiere que PrP Sc sea un catalizador extraordinariamente efectivo, aumentando la velocidad de la reacción de conversión en un factor de alrededor de 10 15 . [59] Este problema no surge si PrP Sc existe solo en formas agregadas como amiloide , donde la cooperatividad puede actuar como una barrera para la conversión espontánea. Es más, a pesar de un esfuerzo considerable, nunca se ha aislado PrP Sc monomérica infecciosa . [60]
Un modelo alternativo supone que la PrP Sc existe sólo en forma de fibrillas y que los extremos de las fibrillas se unen a la PrP C y la convierten en PrP Sc . Si esto fuera todo, la cantidad de priones aumentaría linealmente , formando fibrillas cada vez más largas. Pero durante la enfermedad priónica se observa un crecimiento exponencial tanto de la PrP Sc como de la cantidad de partículas infecciosas . [61] [62] [63] Esto se puede explicar teniendo en cuenta la rotura de fibrillas. [64 ] Se ha encontrado una solución matemática para la tasa de crecimiento exponencial resultante de la combinación del crecimiento de fibrillas y la rotura de fibrillas. [65] La tasa de crecimiento exponencial depende en gran medida de la raíz cuadrada de la concentración de PrP C. [65] El período de incubación está determinado por la tasa de crecimiento exponencial, y los datos in vivo sobre enfermedades priónicas en ratones transgénicos coinciden con esta predicción. [65] La misma dependencia de la raíz cuadrada también se observa in vitro en experimentos con una variedad de diferentes proteínas amiloides . [66]
El mecanismo de replicación de los priones tiene implicaciones para el diseño de fármacos. Dado que el período de incubación de las enfermedades priónicas es tan largo, un fármaco eficaz no necesita eliminar todos los priones, sino simplemente reducir la velocidad de crecimiento exponencial. Los modelos predicen que la forma más eficaz de lograrlo, utilizando un fármaco con la dosis más baja posible, es encontrar un fármaco que se una a los extremos de las fibrillas y les impida seguir creciendo. [67]
Los investigadores del Dartmouth College descubrieron que las moléculas cofactoras endógenas del huésped, como la molécula de fosfolípidos (por ejemplo, la fosfatidiletanolamina) y los polianiones (por ejemplo, las moléculas de ARN monocatenario) son necesarias para formar moléculas de PrP Sc con altos niveles de infectividad específica in vitro , mientras que las moléculas de PrP Sc de solo proteína parecen carecer de niveles significativos de infectividad biológica. [68] [69]
Muchas especies de mamíferos diferentes pueden verse afectadas por enfermedades priónicas, ya que la proteína priónica (PrP) es muy similar en todos los mamíferos. [80] Debido a las pequeñas diferencias en la PrP entre diferentes especies, es inusual que una enfermedad priónica se transmita de una especie a otra. Sin embargo, se cree que la variante humana de la enfermedad priónica, la enfermedad de Creutzfeldt-Jakob, es causada por un prión que normalmente infecta al ganado, causando encefalopatía espongiforme bovina y se transmite a través de la carne infectada. [81]
Todas las enfermedades priónicas conocidas son intratables y fatales. [9] [82] [83]
Hasta 2015, se consideraba que todas las enfermedades priónicas de mamíferos conocidas eran causadas por la proteína priónica PrP ; en 2015, se descubrió que la atrofia multisistémica era transmisible y se planteó la hipótesis de que era causada por un nuevo prión, la forma mal plegada de una proteína llamada alfa -sinucleína . [10] La forma endógena, correctamente plegada de la proteína priónica se denomina PrP C (por Común o Celular ), mientras que la forma mal plegada vinculada a la enfermedad se denomina PrP Sc (por Scrapie ), en honor a una de las enfermedades vinculadas por primera vez a los priones y la neurodegeneración. [ 26] [11] No se conoce la estructura precisa del prión, aunque se pueden formar espontáneamente combinando PrP C , ácido poliadenílico homopolimérico y lípidos en una reacción de amplificación cíclica de plegamiento incorrecto de proteínas (PMCA) incluso en ausencia de priones infecciosos preexistentes. [68] Este resultado es una prueba más de que la replicación de priones no requiere información genética. [84]
Transmisión
Se ha reconocido que las enfermedades priónicas pueden surgir de tres formas diferentes: adquiridas, familiares o esporádicas. [85] A menudo se supone que la forma enferma interactúa directamente con la forma normal para hacer que reorganice su estructura. Una idea, la hipótesis de la "proteína X", es que una proteína celular aún no identificada (proteína X) permite la conversión de PrP C en PrP Sc al unir una molécula de cada una de las dos en un complejo. [86]
El principal método de infección en los animales es la ingestión. Se cree que los priones pueden depositarse en el medio ambiente a través de los restos de animales muertos y de la orina, la saliva y otros fluidos corporales. Luego pueden permanecer en el suelo al unirse a la arcilla y otros minerales. [87]
Un equipo de investigación de la Universidad de California ha aportado pruebas de la teoría de que la infección puede producirse a partir de priones presentes en el estiércol. [88] Y, puesto que el estiércol está presente en muchas zonas que rodean los embalses de agua, así como se utiliza en muchos campos de cultivo, plantea la posibilidad de una transmisión generalizada. Aunque se informó inicialmente en enero de 2011 de que los investigadores habían descubierto que los priones se propagaban a través de la transmisión aérea en partículas de aerosol en un experimento de pruebas con animales centrado en la infección por scrapie en ratones de laboratorio , [89] este informe se retractó en 2024. [90] En 2011 se publicó una prueba preliminar que apoyaba la idea de que los priones pueden transmitirse a través del uso de gonadotropina menopáusica humana derivada de la orina , administrada para el tratamiento de la infertilidad . [91]
Priones en plantas
En 2015, investigadores del Centro de Ciencias de la Salud de la Universidad de Texas en Houston descubrieron que las plantas pueden ser un vector de priones. Cuando los investigadores alimentaron a hámsteres con hierba que crecía en el suelo donde estaba enterrado un ciervo que murió de enfermedad crónica debilitante (CWD), los hámsteres enfermaron de CWD, lo que sugiere que los priones pueden unirse a las plantas, que luego los llevan a la estructura de las hojas y los tallos, donde pueden ser comidos por los herbívoros, completando así el ciclo. Por lo tanto, es posible que haya una cantidad progresivamente acumulada de priones en el medio ambiente. [92] [93]
Esterilización
Las partículas infecciosas que poseen ácido nucleico dependen de él para dirigir su replicación continua. Los priones, sin embargo, son infecciosos por su efecto sobre las versiones normales de la proteína. Por lo tanto, la esterilización de los priones requiere la desnaturalización de la proteína a un estado en el que la molécula ya no puede inducir el plegamiento anormal de las proteínas normales. En general, los priones son bastante resistentes a las proteasas , el calor, la radiación ionizante y los tratamientos con formaldehído , [94] aunque su infectividad puede reducirse con dichos tratamientos. La descontaminación eficaz de priones depende de la hidrólisis de proteínas o la reducción o destrucción de la estructura terciaria de la proteína . Los ejemplos incluyen hipoclorito de sodio , hidróxido de sodio y detergentes fuertemente ácidos como LpH. [95]
La Organización Mundial de la Salud recomienda cualquiera de los tres procedimientos siguientes para la esterilización de todos los instrumentos quirúrgicos resistentes al calor para garantizar que no estén contaminados con priones:
Sumergir en hipoclorito de sodio 1N (20.000 partes por millón de cloro disponible) durante 1 hora; transferir los instrumentos al agua; calentar en un autoclave de desplazamiento por gravedad a 121 °C durante 1 hora; limpiar; y luego realizar los procesos de esterilización de rutina.
Sumergir en hidróxido de sodio 1N o hipoclorito de sodio (20.000 partes por millón de cloro disponible) durante 1 hora; retirar y enjuagar con agua, luego transferir a una cacerola abierta y calentar en un autoclave de desplazamiento por gravedad (121 °C) o en un autoclave de carga porosa (134 °C) durante 1 hora; limpiar; y luego realizar procesos de esterilización de rutina. [96]
Se ha descubierto que la esterilización a 134 °C (273 °F) durante 18 minutos en un autoclave de vapor presurizado es algo eficaz para desactivar el agente de la enfermedad. [97] [98] Actualmente se está estudiando la esterilización con ozono como un método potencial para la desnaturalización y desactivación de priones. [99] Otros enfoques que se están desarrollando incluyen el tratamiento con tiourea - urea , el tratamiento con cloruro de guanidinio , [100] y la subtilisina especial resistente al calor combinada con calor y detergente. [101] Un método suficiente para esterilizar priones en un material puede fallar en otro. [102]
Aún no se ha logrado la renaturalización de un prión completamente desnaturalizado a un estado infeccioso; sin embargo, los priones parcialmente desnaturalizados pueden renaturalizarse a un estado infeccioso bajo ciertas condiciones artificiales. [103]
Resistencia a la degradación en la naturaleza
Hay pruebas abrumadoras de que los priones resisten la degradación y persisten en el medio ambiente durante años, y las proteasas no los degradan. La evidencia experimental muestra que los priones no ligados se degradan con el tiempo, mientras que los priones ligados al suelo permanecen en niveles estables o en aumento, lo que sugiere que es probable que los priones se acumulen en el medio ambiente. [104] [105] Un estudio de 2015 realizado por científicos estadounidenses descubrió que el secado y la humectación repetidos pueden hacer que los priones ligados al suelo sean menos infecciosos, aunque esto dependía del tipo de suelo al que estaban ligados. [106]
Degradación por seres vivos
Estudios más recientes sugieren que los priones de scrapie pueden ser degradados por diversos mecanismos celulares. La inhibición de la autofagia acelera la acumulación de priones mientras que el estímulo de la autofagia promueve la eliminación de priones. [107] El sistema de proteosoma ubiquitina parece ser capaz de degradar agregados suficientemente pequeños. [107] Además, se ha descubierto que la queratinasa de B. licheniformis , [108] [109] la serina proteasa alcalina de Streptomyces sp , [110] la pernisina similar a la subtilisina de Aeropyrum pernix , [111] la proteasa alcalina de Nocardiopsis sp , [112] la nattoquinasa de B. subtilis , [113] las subtilisinas diseñadas de B. lentus [114] [115] y la serina proteasa de tres especies de líquenes [116] degradan PrP Sc .
Hongos
Las proteínas que muestran un comportamiento de tipo priónico también se encuentran en algunos hongos , lo que ha sido útil para ayudar a comprender los priones de los mamíferos. Los priones fúngicos no siempre causan enfermedades en sus huéspedes. [117] En la levadura, el replegamiento de proteínas a la configuración priónica es asistido por proteínas chaperonas como Hsp104 . [118] Todos los priones conocidos inducen la formación de un pliegue amiloide , en el que la proteína se polimeriza en un agregado que consiste en láminas beta muy compactas . Los agregados amiloides son fibrillas, que crecen en sus extremos y se replican cuando la rotura hace que dos extremos en crecimiento se conviertan en cuatro extremos en crecimiento. El período de incubación de las enfermedades priónicas está determinado por la tasa de crecimiento exponencial asociada con la replicación priónica, que es un equilibrio entre el crecimiento lineal y la rotura de agregados. [65]
Las proteínas fúngicas que exhiben un cambio conformacional modelado [ se necesita más explicación ] fueron descubiertas en la levadura Saccharomyces cerevisiae por Reed Wickner a principios de la década de 1990. Por su similitud mecanística con los priones de los mamíferos, se las denominó priones de levadura . Posteriormente, también se encontró un prión en el hongo Podospora anserina . Estos priones se comportan de manera similar a la PrP, pero, en general, no son tóxicos para sus huéspedes. El grupo de Susan Lindquist en el Instituto Whitehead ha argumentado que algunos de los priones fúngicos no están asociados con ningún estado patológico, pero pueden tener un papel útil; sin embargo, los investigadores del NIH también han proporcionado argumentos que sugieren que los priones fúngicos podrían considerarse un estado patológico. [119] Existe evidencia de que las proteínas fúngicas han desarrollado funciones específicas que son beneficiosas para el microorganismo y que mejoran su capacidad de adaptarse a sus diversos entornos. [120] Además, dentro de las levaduras, los priones pueden actuar como vectores de herencia epigenética , transfiriendo rasgos a la descendencia sin ningún cambio genómico . [121] [122]
La investigación sobre los priones fúngicos ha dado un fuerte apoyo al concepto de proteína únicamente, ya que se ha demostrado que la proteína purificada extraída de células con un estado priónico convierte la forma normal de la proteína en una forma mal plegada in vitro y, en el proceso, preserva la información correspondiente a diferentes cepas del estado priónico. También ha arrojado algo de luz sobre los dominios priónicos, que son regiones en una proteína que promueven la conversión en un prión. Los priones fúngicos han ayudado a sugerir mecanismos de conversión que pueden aplicarse a todos los priones, aunque los priones fúngicos parecen distintos de los priones infecciosos de mamíferos en la falta de cofactor necesario para la propagación. Los dominios priónicos característicos pueden variar entre especies; por ejemplo, los dominios priónicos fúngicos característicos no se encuentran en los priones de mamíferos. [ cita requerida ]
Tratos
No existen tratamientos efectivos para las enfermedades priónicas. [124] Los ensayos clínicos en humanos no han tenido éxito y se han visto obstaculizados por la rareza de las enfermedades priónicas. [124] Aunque algunos tratamientos potenciales han demostrado ser prometedores en el laboratorio, ninguno ha sido efectivo una vez que la enfermedad ha comenzado. [125]
La definición de un dominio priónico surge del estudio de los priones fúngicos. En la levadura, las proteínas prionogénicas tienen un dominio priónico portátil que es necesario y suficiente para la autoformación y la agregación de proteínas. Esto se ha demostrado uniendo el dominio priónico a una proteína reportera, que luego se agrega como un prión conocido. De manera similar, la eliminación del dominio priónico de una proteína priónica fúngica inhibe la prionogénesis. Esta visión modular del comportamiento priónico ha llevado a la hipótesis de que existen dominios priónicos similares en las proteínas animales, además de PrP. [126] Estos dominios priónicos fúngicos tienen varias características de secuencia características. Por lo general, están enriquecidos en residuos de asparagina, glutamina, tirosina y glicina, y un sesgo de asparagina es particularmente propicio para la propiedad agregativa de los priones. Históricamente, la prionogénesis se ha considerado independiente de la secuencia y solo dependiente del contenido relativo de residuos. Sin embargo, se ha demostrado que esto es falso, ya que se ha demostrado que el espaciamiento de las prolinas y los residuos cargados es fundamental para la formación de amiloide. [130]
Los análisis bioinformáticos han predicho que más de 250 proteínas humanas contienen dominios similares a priones (PrLD). Se ha planteado la hipótesis de que estos dominios tienen las mismas propiedades amiloidogénicas y transmisibles que la PrP y las proteínas fúngicas conocidas. Al igual que en la levadura, las proteínas implicadas en la expresión génica y la unión del ARN parecen estar particularmente enriquecidas en los dominios similares a priones, en comparación con otras clases de proteínas. En particular, 29 de las 210 proteínas conocidas con un motivo de reconocimiento de ARN también tienen un supuesto dominio priónico. Mientras tanto, varias de estas proteínas de unión al ARN se han identificado de forma independiente como patógenas en casos de ELA, FTLD-U, enfermedad de Alzheimer y enfermedad de Huntington. [131]
Papel en las enfermedades neurodegenerativas
Se ha planteado la hipótesis de que la patogenicidad de los priones y las proteínas con dominios similares a los priones surge de su capacidad de autogeneración y del crecimiento exponencial resultante de las fibrillas amiloides. La presencia de fibrillas amiloides en pacientes con enfermedades degenerativas ha sido bien documentada. Estas fibrillas amiloides se consideran el resultado de proteínas patógenas que se autopropagan y forman agregados no funcionales altamente estables. [131] Si bien esto no implica necesariamente una relación causal entre las enfermedades amiloides y las degenerativas, la toxicidad de ciertas formas amiloides y la sobreproducción de amiloide en casos familiares de trastornos degenerativos respaldan la idea de que la formación de amiloide es generalmente tóxica. [132]
En concreto, se ha encontrado agregación de TDP-43 , una proteína de unión al ARN, en pacientes con ELA/EMN, y se han identificado mutaciones en los genes que codifican estas proteínas en casos familiares de ELA/EMN. Estas mutaciones promueven el plegamiento incorrecto de las proteínas en una conformación similar a la de un prión. La forma mal plegada de TDP-43 forma inclusiones citoplasmáticas en las neuronas afectadas y se encuentra agotada en el núcleo. Además de la ELA/EMN y la DLFT-U, la patología de TDP-43 es una característica de muchos casos de enfermedad de Alzheimer, enfermedad de Parkinson y enfermedad de Huntington. El plegamiento incorrecto de TDP-43 está dirigido en gran medida por su dominio similar al prión. Este dominio es inherentemente propenso al plegamiento incorrecto, mientras que se ha descubierto que las mutaciones patológicas en TDP-43 aumentan esta propensión al plegamiento incorrecto, lo que explica la presencia de estas mutaciones en casos familiares de ELA/EMN. Al igual que en la levadura, se ha demostrado que el dominio priónico de TDP-43 es necesario y suficiente para el plegamiento incorrecto y la agregación de proteínas. [126]
De manera similar, se han identificado mutaciones patógenas en los dominios priónicos de las riboproteínas nucleares heterogéneas hnRNPA2B1 y hnRNPA1 en casos familiares de degeneración de neuronas motoras, musculares, cerebrales y óseas. La forma salvaje de todas estas proteínas muestra una tendencia a autoensamblarse en fibrillas amiloides, mientras que las mutaciones patógenas exacerban este comportamiento y conducen a una acumulación excesiva. [133]
Armamentización
En teoría, los priones podrían emplearse como un agente armado . [134] [135] Con tasas de mortalidad potenciales del 100%, los priones podrían ser un arma biológica eficaz, a veces llamada "arma bioquímica", porque un prión es un bioquímico. Un aspecto desfavorable es que los períodos de incubación de los priones son muy largos. La exposición intensa y persistente a priones en el intestino podría acortar el inicio general. [136] Otro aspecto del uso de priones en la guerra es la dificultad de detección y descontaminación . [137]
Historia
En los siglos XVIII y XIX, se observó que la exportación de ovejas desde España coincidía con una enfermedad llamada scrapie . Esta enfermedad hacía que los animales afectados "se tumbaran, se mordieran los pies y las patas, se frotaran el lomo contra los postes, no prosperaran, dejaran de alimentarse y finalmente quedaran cojos" . [138] También se observó que la enfermedad tenía un largo período de incubación que es una característica clave de las encefalopatías espongiformes transmisibles (EET) . Aunque en aquel entonces no se conocía la causa del scrapie, es probable que sea la primera encefalopatía espongiforme transmisible registrada. [139]
En la década de 1950, Carleton Gajdusek comenzó una investigación que finalmente demostró que el kuru podía transmitirse a los chimpancés por lo que posiblemente era un nuevo agente infeccioso, trabajo por el que finalmente ganó el premio Nobel de 1976. Durante la década de 1960, dos investigadores con sede en Londres, la bióloga de radiación Tikvah Alper y el biofísico John Stanley Griffith , desarrollaron la hipótesis de que las encefalopatías espongiformes transmisibles son causadas por un agente infeccioso que consiste únicamente en proteínas. [140] [141] Investigaciones anteriores de EJ Field sobre el scrapie y el kuru habían encontrado evidencia de la transferencia de polisacáridos patológicamente inertes que solo se vuelven infecciosos después de la transferencia, en el nuevo huésped. [142] [143] Alper y Griffith querían explicar el descubrimiento de que el misterioso agente infeccioso que causaba las enfermedades scrapie y Creutzfeldt-Jakob resistía la radiación ionizante . [144] Griffith propuso tres formas en las que una proteína podría ser un patógeno . [145]
En la primera hipótesis , sugirió que si la proteína es el producto de un gen normalmente suprimido , y la introducción de la proteína podría inducir la expresión del gen, es decir, despertar el gen latente, entonces el resultado sería un proceso indistinguible de la replicación, ya que la expresión del gen produciría la proteína, que luego despertaría el gen en otras células . [ cita requerida ]
Su segunda hipótesis constituye la base de la teoría moderna del prión y propone que una forma anormal de una proteína celular puede convertir proteínas normales del mismo tipo en su forma anormal, lo que conduce a la replicación. [ cita requerida ]
Su tercera hipótesis proponía que el agente podría ser un anticuerpo si el anticuerpo fuera su propio antígeno diana , ya que un anticuerpo de este tipo daría lugar a que se produjeran cada vez más anticuerpos contra sí mismo. Sin embargo, Griffith reconoció que era poco probable que esta tercera hipótesis fuera cierta debido a la falta de una respuesta inmunitaria detectable . [146]
Francis Crick reconoció la importancia potencial de la hipótesis de la proteína exclusiva de Griffith para la propagación del scrapie en la segunda edición de su " Dogma central de la biología molecular " (1970): mientras afirmaba que el flujo de información de secuencia de proteína a proteína, o de proteína a ARN y ADN estaba "impedido", señaló que la hipótesis de Griffith era una contradicción potencial (aunque Griffith no la promovió de esa manera). [147] La hipótesis revisada se formuló más tarde, en parte, para dar cabida a la transcripción inversa (que tanto Howard Temin como David Baltimore descubrieron en 1970). [148]
En 1982, Stanley B. Prusiner, de la Universidad de California en San Francisco , anunció que su equipo había purificado la proteína infecciosa hipotética, que no parecía estar presente en huéspedes sanos, aunque no lograron aislar la proteína hasta dos años después del anuncio de Prusiner. [149] [14] La proteína fue denominada prión, por "partícula infecciosa proteínica", derivada de las palabras proteína e infección . Cuando se descubrió el prión, la primera hipótesis de Griffith, de que la proteína era el producto de un gen normalmente silencioso, fue apoyada por muchos. Sin embargo, posteriormente se descubrió que la misma proteína existe en huéspedes normales, pero en una forma diferente. [150]
Tras el descubrimiento de la misma proteína en una forma diferente en individuos no infectados, la proteína específica de la que estaba compuesto el prión se denominó proteína priónica (PrP), y la segunda hipótesis de Griffith de que una forma anormal de una proteína huésped puede convertir otras proteínas del mismo tipo en su forma anormal, se convirtió en la teoría dominante. [146] Prusiner recibió el Premio Nobel de Fisiología o Medicina en 1997 por su investigación sobre los priones. [151] [152]
^ "Pronunciación inglesa de prion". Diccionario Cambridge . Cambridge University Press. Archivado desde el original el 24 de abril de 2017 . Consultado el 30 de marzo de 2020 .
^ "Definición de prión". Dictionary.com . Random House, Inc. 2021. Definición 2 de 2. Archivado desde el original el 12 de septiembre de 2021 . Consultado el 12 de septiembre de 2021 .
^ "Encefalopatías espongiformes transmisibles". Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares . Consultado el 23 de abril de 2023 .
^ "Enfermedades priónicas". Enfermedades y afecciones. Instituto Nacional de Salud. Archivado desde el original el 22 de mayo de 2020. Consultado el 20 de junio de 2018 .
^ Kumar V (2021). Robbins & Cotran Base patológica de la enfermedad (10.ª ed.).
^ "¿Qué es un prión?". Scientific American . Archivado desde el original el 16 de mayo de 2018. Consultado el 15 de mayo de 2018 .
^ "Prion infectious agent". Encyclopaedia Britannica. Archivado desde el original el 16 de mayo de 2018. Consultado el 15 de mayo de 2018 .
^ Prusiner SB (junio de 1991). "Biología molecular de las enfermedades priónicas". Science . 252 (5012): 1515–1522. Bibcode :1991Sci...252.1515P. doi :10.1126/science.1675487. PMID 1675487. S2CID 22417182.
^ ab Prusiner SB (noviembre de 1998). "Priones". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 95 (23): 13363–13383. Bibcode :1998PNAS...9513363P. doi : 10.1073/pnas.95.23.13363 . PMC 33918 . PMID 9811807.
^ abc Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al. (septiembre de 2015). "Evidencia de que los priones de α-sinucleína causan atrofia multisistémica en humanos con parkinsonismo". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 112 (38): E5308–E5317. Bibcode :2015PNAS..112E5308P. doi : 10.1073/pnas.1514475112 . PMC 4586853 . PMID 26324905. Resumen para legos: Makin S (1 de septiembre de 2015). "Una señal de alerta de una enfermedad neurodegenerativa que puede ser transmisible". Scientific American .
^ ab Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (febrero de 2009). "La proteína priónica celular media el deterioro de la plasticidad sináptica por oligómeros de beta-amiloide". Nature . 457 (7233): 1128–1132. Bibcode :2009Natur.457.1128L. doi :10.1038/nature07761. PMC 2748841 . PMID 19242475.
^ ab Olanow CW, Brundin P (enero de 2013). "Enfermedad de Parkinson y alfa-sinucleína: ¿es la enfermedad de Parkinson un trastorno similar a los priones?". Trastornos del movimiento . 28 (1): 31–40. doi :10.1002/mds.25373. PMID 23390095. S2CID 38287298.
^ Dobson CM (febrero de 2001). "La base estructural del plegamiento de proteínas y sus vínculos con las enfermedades humanas". Philosophical Transactions of the Royal Society of London. Serie B, Ciencias Biológicas . 356 (1406): 133–145. doi :10.1098/rstb.2000.0758. PMC 1088418 . PMID 11260793.
^ abc Prusiner SB (abril de 1982). «Nuevas partículas infecciosas proteínicas causan scrapie» (PDF) . Science . 216 (4542): 136–144. Bibcode :1982Sci...216..136P. doi :10.1126/science.6801762. PMID 6801762. S2CID 7447120. Archivado desde el original (PDF) el 20 de julio de 2020.
^ "Stanley B. Prusiner – Autobiografía". NobelPrize.org. Archivado desde el original el 16 de junio de 2013. Consultado el 2 de enero de 2007 .
^ "Dorland's Illustrated Medical Dictionary". Elsevier. Archivado desde el original el 11 de enero de 2014. Consultado el 22 de julio de 2016 .
^ ab «Diccionario íntegro de Merriam-Webster». Merriam-Webster. Archivado desde el original el 25 de mayo de 2020. Consultado el 22 de julio de 2016 .
^ "The American Heritage Dictionary of the English Language". Houghton Mifflin Harcourt. Archivado desde el original el 25 de septiembre de 2015. Consultado el 22 de julio de 2016 .
^ Priola SA, Chesebro B, Caughey B (mayo de 2003). "Biomedicina. Una visión desde arriba: enfermedades priónicas desde 10 000 pies". Science . 300 (5621): 917–919. doi :10.1126/science.1085920. PMID 12738843. S2CID 38459669. Archivado desde el original el 28 de julio de 2020 . Consultado el 28 de julio de 2020 .
^ Artikis E, Kraus A, Caughey B (agosto de 2022). "Biología estructural de priones de mamíferos ex vivo". The Journal of Biological Chemistry . 298 (8): 102181. doi : 10.1016/j.jbc.2022.102181 . PMC 9293645 . PMID 35752366.
^ Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, McNicol A (mayo de 2006). "La proteína priónica celular se libera en los exosomas de las plaquetas activadas". Blood . 107 (10): 3907–3911. doi : 10.1182/blood-2005-02-0802 . PMID 16434486. S2CID 34141310.
^ Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K (agosto de 1997). "Caracterización por RMN de la proteína priónica murina recombinante de longitud completa, mPrP(23-231)" (PDF) . FEBS Letters . 413 (2): 282–288. Bibcode :1997FEBSL.413..282R. doi :10.1016/S0014-5793(97)00920-4. PMID 9280298. S2CID 39791520.
^ Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, et al. (diciembre de 1997). "Estructura de la proteína priónica de hámster recombinante de longitud completa PrP(29-231): el extremo N es muy flexible". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 94 (25): 13452–13457. Bibcode :1997PNAS...9413452D. doi : 10.1073/pnas.94.25.13452 . PMC 28326 . PMID 9391046.
^ Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, et al. (febrero de 1998). "Una forma transmembrana de la proteína priónica en enfermedades neurodegenerativas" (PDF) . Science . 279 (5352): 827–834. Bibcode :1998Sci...279..827H. doi :10.1126/science.279.5352.827. PMID 9452375. S2CID 20176119. Archivado desde el original (PDF) el 23 de febrero de 2019.
^ abc Carp RI, Kascap RJ (2004). "Apuntando a los agentes infecciosos de la encefalopatía espongiforme transmisible". En Krull IS, Nunnally BK (eds.). Priones y enfermedad de las vacas locas . Nueva York: Marcel Dekker. p. 6. ISBN978-0-8247-4083-2Archivado del original el 20 de agosto de 2020 . Consultado el 2 de junio de 2020 .
^ Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. (1997). "La proteína priónica celular se une al cobre in vivo". Nature . 390 (6661): 684–687. Bibcode :1997Natur.390..684B. doi :10.1038/37783. PMID 9414160. S2CID 4388803.
^ Weissmann C (noviembre de 2004). "El estado del prión". Nature Reviews. Microbiology . 2 (11): 861–871. doi :10.1038/nrmicro1025. PMID 15494743. S2CID 20992257.
^ Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, et al. (marzo de 2009). Weissmann C (ed.). "Regulación de la adhesión celular embrionaria por la proteína priónica". PLOS Biology . 7 (3): e55. doi : 10.1371/journal.pbio.1000055 . PMC 2653553 . PMID 19278297.
^ Liebert A, Bicknell B, Adams R (2014). "Señalización de proteínas priónicas en el sistema nervioso: una revisión y perspectiva". Signal Transduction Insights . 3 : STI.S12319. doi : 10.4137/STI.S12319 . ISSN 1178-6434.
^ Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS (agosto de 1991). "Análisis de la estructura secundaria de la proteína asociada a la tembladera PrP 27-30 en agua mediante espectroscopia infrarroja". Bioquímica . 30 (31): 7672–7680. doi :10.1021/bi00245a003. PMID 1678278.
^ Safar J, Roller PP, Gajdusek DC, Gibbs CJ (septiembre de 1993). "Transiciones conformacionales, disociación y desdoblamiento de la proteína amiloide (prión) del scrapie". The Journal of Biological Chemistry . 268 (27): 20276–20284. doi : 10.1016/s0021-9258(20)80725-x . PMID 8104185.
^ Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. (diciembre de 1993). "La conversión de hélices alfa en láminas beta es una característica de la formación de las proteínas priónicas del scrapie". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 90 (23): 10962–10966. Bibcode :1993PNAS...9010962P. doi : 10.1073/pnas.90.23.10962 . PMC 47901 . PMID 7902575.
^ Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, et al. (noviembre de 2021). "Comparación de estructura y cepas de alta resolución de priones infecciosos de mamíferos". Molecular Cell . 81 (21): 4540–4551.e6. doi :10.1016/j.molcel.2021.08.011. PMID 34433091.
^ Hoyt F, Standke HG, Artikis E, Schwartz CL, Hansen B, Li K, et al. (julio de 2022). "La estructura crio-EM del prión RML sin anclaje revela variaciones en los motivos compartidos entre distintas cepas". Nature Communications . 13 (1): 4005. Bibcode :2022NatCo..13.4005H. doi :10.1038/s41467-022-30458-6. PMC 9279418 . PMID 35831291.
^ Manka SW, Zhang W, Wenborn A, Betts J, Joiner S, Saibil HR, et al. (julio de 2022). "Estructura crio-EM de 2,7 Å de fibrillas priónicas RML ex vivo". Nature Communications . 13 (1): 4004. Bibcode :2022NatCo..13.4004M. doi :10.1038/s41467-022-30457-7. PMC 9279362 . PMID 35831275.
^ Hallinan GI, Ozcan KA, Hoq MR, Cracco L, Vago FS, Bharath SR, et al. (septiembre de 2022). "Estructuras crio-EM de filamentos de proteína priónica de la enfermedad de Gerstmann-Sträussler-Scheinker". Acta Neuropathologica . 144 (3): 509–520. doi :10.1007/s00401-022-02461-0. PMC 9381446 . PMID 35819518.
^ Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, Cohen FE, et al. (marzo de 2002). "Estudios estructurales de la proteína priónica del scrapie mediante cristalografía electrónica". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 99 (6): 3563–3568. Bibcode :2002PNAS...99.3563W. doi : 10.1073/pnas.052703499 . PMC 122563 . PMID 11891310.
^ Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, et al. (noviembre de 2021). "Comparación de estructura y cepas de alta resolución de priones infecciosos de mamíferos". Molecular Cell . 81 (21): 4540–4551.e6. doi :10.1016/j.molcel.2021.08.011. PMID 34433091.
^ Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B (junio de 1995). "Propagación no genética de propiedades específicas de cepas de la proteína priónica del scrapie". Nature . 375 (6533): 698–700. Bibcode :1995Natur.375..698B. doi :10.1038/375698a0. PMID 7791905. S2CID 4355092.
^ Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. (diciembre de 1996). "Evidencia de la conformación de la isoforma patológica de la proteína priónica que codifica y propaga la diversidad priónica". Science . 274 (5295): 2079–2082. Bibcode :1996Sci...274.2079T. doi :10.1126/science.274.5295.2079. PMID 8953038.
^ Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. (octubre de 1998). "Ocho cepas de priones tienen moléculas de PrP(Sc) con diferentes conformaciones". Nature Medicine . 4 (10): 1157–1165. doi :10.1038/2654. PMID 9771749. S2CID 6031488.
^ Hoyt F, Alam P, Artikis E, Schwartz CL, Hughson AG, Race B, et al. (noviembre de 2022). "La crio-EM de cepas de priones del mismo genotipo de huésped identifica determinantes conformacionales". PLOS Pathogens . 18 (11): e1010947. doi : 10.1371/journal.ppat.1010947 . PMC 9671466 . PMID 36342968.
^ Manka SW, Wenborn A, Betts J, Joiner S, Saibil HR, Collinge J, et al. (mayo de 2023). "Una base estructural para la diversidad de cepas de priones". Nature Chemical Biology . 19 (5): 607–613. doi :10.1038/s41589-022-01229-7. PMC 10154210 . PMID 36646960.
^ Kurt TD, Sigurdson CJ (2016). "Transmisión entre especies de priones de CWD". Prion . 10 (1): 83–91. doi :10.1080/19336896.2015.1118603. PMC 4981193 . PMID 26809254.
^ Riesner D (junio de 2003). "Bioquímica y estructura de PrP(C) y PrP(Sc)". British Medical Bulletin . 66 (1): 21–33. doi : 10.1093/bmb/66.1.21 . PMID 14522846.
^ Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. (agosto de 1994). "Formación de proteína priónica resistente a proteasas sin células". Nature . 370 (6489): 471–474. Bibcode :1994Natur.370..471K. doi :10.1038/370471a0. hdl : 1721.1/42578 . PMID 7913989. S2CID 4337709.
^ Saborio GP, Permanne B, Soto C (junio de 2001). "Detección sensible de la proteína priónica patológica mediante amplificación cíclica del plegamiento incorrecto de proteínas". Nature . 411 (6839): 810–813. Bibcode :2001Natur.411..810S. doi :10.1038/35081095. PMID 11459061. S2CID 4317585.
^ Bieschke J, Weber P, Sarafoff N, Beekes M, Giese A, Kretzschmar H (agosto de 2004). "Autopropagación autocatalítica de proteína priónica mal plegada". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 101 (33): 12207–12211. Bibcode :2004PNAS..10112207B. doi : 10.1073/pnas.0404650101 . PMC 514458 . PMID 15297610.
^ Abbott A (24 de enero de 2010). "Los priones sanos protegen los nervios". Nature . doi :10.1038/news.2010.29. S2CID 84980140.
^ Nailwal H, Chan FK (enero de 2019). "Necroptosis en la inflamación antiviral". Muerte celular y diferenciación . 26 (1): 4–13. doi :10.1038/s41418-018-0172-x. PMC 6294789. PMID 30050058 .
^ Shorter J, Lindquist S (junio de 2005). "Priones como conductos adaptativos de la memoria y la herencia". Nature Reviews. Genetics . 6 (6): 435–450. doi :10.1038/nrg1616. PMID 15931169. S2CID 5575951.
^ Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA (noviembre de 2004). "Plasticidad sináptica del hipocampo en ratones desprovistos de proteína priónica celular". Investigación cerebral. Investigación cerebral molecular . 131 (1–2): 58–64. doi :10.1016/j.molbrainres.2004.08.004. PMID 15530652.
^ Caiati MD, Safiulina VF, Fattorini G, Sivakumaran S, Legname G, Cherubini E (febrero de 2013). "PrPC controla a través de la proteína quinasa A la dirección de la plasticidad sináptica en el hipocampo inmaduro". The Journal of Neuroscience . 33 (7): 2973–2983. doi :10.1523/JNEUROSCI.4149-12.2013. PMC 6619229 . PMID 23407955.
^ Sudhakaran IP, Ramaswami M (mayo de 2017). "Consolidación de la memoria a largo plazo: el papel de las proteínas de unión al ARN con dominios similares a priones". RNA Biology . 14 (5): 568–586. doi :10.1080/15476286.2016.1244588. PMC 5449092 . PMID 27726526.
^ Zhang CC, Steele AD, Lindquist S, Lodish HF (febrero de 2006). "La proteína priónica se expresa en células madre hematopoyéticas que se repoblaron a largo plazo y es importante para su autorrenovación". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 103 (7): 2184–2189. Bibcode :2006PNAS..103.2184Z. doi : 10.1073/pnas.0510577103 . PMC 1413720 . PMID 16467153.
^ Lathe R, Darlix JL (diciembre de 2017). "Proteína priónica PRNP: ¿Un nuevo actor en la inmunidad innata? La conexión Aβ". Journal of Alzheimer's Disease Reports . 1 (1): 263–275. doi :10.3233/ADR-170037. PMC 6159716 . PMID 30480243.
^ Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB (abril de 1994). "Pistas estructurales de la replicación de priones". Science . 264 (5158): 530–531. Bibcode :1994Sci...264..530C. doi :10.1126/science.7909169. PMID 7909169.
^ Eigen M (diciembre de 1996). "Priónica o la base cinética de las enfermedades priónicas". Química biofísica . 63 (1): A1-18. doi :10.1016/S0301-4622(96)02250-8. PMID 8981746.
^ Vázquez-Fernández E, Young HS, Requena JR, Wille H (2017). "La estructura de los priones de mamíferos y sus agregados". Revista Internacional de Biología Celular y Molecular . 329 : 277–301. doi :10.1016/bs.ircmb.2016.08.013. ISBN978-0-12-812251-8. Número de identificación personal 28109330.
^ Bolton DC, Rudelli RD, Currie JR, Bendheim PE (diciembre de 1991). "Copurificación de Sp33-37 y agente de scrapie del cerebro de hámster antes de la histopatología detectable y la enfermedad clínica". The Journal of General Virology . 72 (12): 2905–2913. doi : 10.1099/0022-1317-72-12-2905 . PMID 1684986.
^ Jendroska K, Heinzel FP, Torchia M, Stowring L, Kretzschmar HA, Kon A, et al. (septiembre de 1991). "La acumulación de proteína priónica resistente a la proteinasa en el cerebro del hámster sirio se correlaciona con la patología regional y la infectividad del scrapie". Neurología . 41 (9): 1482–1490. doi :10.1212/WNL.41.9.1482. PMID 1679911. S2CID 13098083.
^ Beekes M, Baldauf E, Diringer H (agosto de 1996). "Aparición secuencial y acumulación de marcadores patognomónicos en el sistema nervioso central de hámsteres infectados por vía oral con scrapie". The Journal of General Virology . 77 (Pt 8) (8): 1925–1934. doi : 10.1099/0022-1317-77-8-1925 . PMID 8760444.
^ Bamborough P, Wille H, Telling GC, Yehiely F, Prusiner SB, Cohen FE (1996). "Estructura de la proteína priónica y replicación de scrapie: investigaciones teóricas, espectroscópicas y genéticas". Simposios de Cold Spring Harbor sobre biología cuantitativa . 61 : 495–509. doi :10.1101/SQB.1996.061.01.050 (inactivo el 10 de julio de 2024). PMID 9246476.{{cite journal}}: CS1 maint: DOI inactivo a partir de julio de 2024 ( enlace )
^ abcd Masel J, Jansen VA, Nowak MA (marzo de 1999). "Cuantificación de los parámetros cinéticos de la replicación priónica". Química biofísica . 77 (2–3): 139–152. CiteSeerX 10.1.1.178.8812 . doi :10.1016/S0301-4622(99)00016-2. PMID 10326247.
^ Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, et al. (diciembre de 2009). "Una solución analítica para la cinética del ensamblaje de filamentos rompibles". Science . 326 (5959): 1533–1537. Bibcode :2009Sci...326.1533K. doi :10.1126/science.1178250. PMID 20007899. S2CID 6267152.
^ Masel J, Jansen VA (diciembre de 2000). "Diseño de fármacos para detener la formación de agregados priónicos y otros amiloides". Química biofísica . 88 (1–3): 47–59. doi :10.1016/S0301-4622(00)00197-6. PMID 11152275.
^ ab Deleault NR, Harris BT, Rees JR, Supattapone S (junio de 2007). "Formación de priones nativos a partir de componentes mínimos in vitro". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 104 (23): 9741–9746. doi : 10.1073/pnas.0702662104 . PMC 1887554 . PMID 17535913.
^ Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, et al. (julio de 2012). "Las moléculas de cofactor mantienen la conformación infecciosa y restringen las propiedades de la cepa en priones purificados". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 109 (28): E1938–E1946. doi : 10.1073/pnas.1206999109 . PMC 3396481 . PMID 22711839.
^ abcdefghi "90. Priones". Índice ICTVdB de virus . Sitio web de los Institutos Nacionales de Salud de EE. UU. 14 de febrero de 2002. Archivado desde el original el 27 de agosto de 2009. Consultado el 28 de febrero de 2010 .
^ Babelhadj B, Di Bari MA, Pirisinu L, Chiappini B, Gaouar SB, Riccardi G, et al. (junio de 2018). "Enfermedad priónica en dromedarios, Argelia". Enfermedades Infecciosas Emergentes . 24 (6): 1029–1036. doi :10.3201/eid2406.172007. PMC 6004840 . PMID 29652245.
^ Hussein MF, Al-Mufarrej SI (2004). "Prion Diseases: A Review; II. Prion Diseases in Man and Animals" (PDF) . Revista científica de la Universidad Rey Faisal (ciencias básicas y aplicadas) . 5 (2): 139. Archivado (PDF) desde el original el 21 de abril de 2016. Consultado el 9 de abril de 2016 .
^ Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, et al. (mayo de 1999). "Conformación de la proteína priónica en un paciente con insomnio fatal esporádico". The New England Journal of Medicine . 340 (21): 1630–1638. doi : 10.1056/NEJM199905273402104 . PMID 10341275. Resumen para legos: "Las proteínas de la EEB pueden causar insomnio mortal". BBC News . 28 de mayo de 1999.
^ Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, et al. (agosto de 1997). "Encefalopatía espongiforme familiar asociada con una nueva mutación del gen de la proteína priónica". Anales de neurología . 42 (2): 138–146. doi :10.1002/ana.410420203. PMID 9266722. S2CID 22600579.
^ Robbins SL, Cotran RS, Kumar V, Collins T, eds. (1999). Base patológica de la enfermedad de Robbins . Filadelfia: Saunders. ISBN072167335X.
^ Belay ED (1999). "Encefalopatías espongiformes transmisibles en humanos". Revisión anual de microbiología . 53 : 283–314. doi :10.1146/annurev.micro.53.1.283. PMID 10547693. S2CID 18648029.
^ "Prion Diseases" (Enfermedades priónicas). Centros para el Control y Prevención de Enfermedades de Estados Unidos. 26 de enero de 2006. Archivado desde el original el 4 de marzo de 2010. Consultado el 28 de febrero de 2010 .
^ Imran M, Mahmood S (diciembre de 2011). "Una descripción general de las enfermedades priónicas humanas". Virology Journal . 8 (1): 559. doi : 10.1186/1743-422X-8-559 . PMC 3296552 . PMID 22196171.
^ Mastrianni JA (abril de 2010). "La genética de las enfermedades priónicas". Genética en Medicina . 12 (4): 187–195. doi : 10.1097/GIM.0b013e3181cd7374 . PMID 20216075.
^ Collinge J (2001). "Prion diseases of humans and animals: their causes and molecular basis" (PDF) . Revisión anual de neurociencia . 24 : 519–550. doi :10.1146/annurev.neuro.24.1.519. PMID 11283320. S2CID 18915904. Archivado desde el original (PDF) el 25 de febrero de 2019.
^ Ironside JW (marzo de 2006). "Variante de la enfermedad de Creutzfeldt-Jakob: riesgo de transmisión por transfusión sanguínea y terapias sanguíneas". Haemophilia . 12 (Supl 1): 8–15, discusión 26–28. doi : 10.1111/j.1365-2516.2006.01195.x . PMID 16445812.
^ Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, et al. (agosto de 2001). "La redireccionación intracelular de la proteína priónica previene la propagación de PrP(Sc) y retrasa la aparición de la enfermedad priónica". The EMBO Journal . 20 (15): 3957–3966. doi :10.1093/emboj/20.15.3957. PMC 149175 . PMID 11483499.
^ Agarwal A, Mukhopadhyay S (enero de 2022). "Biología de las proteínas priónicas a través de la lente de la separación de fases líquido-líquido". Journal of Molecular Biology . 434 (1): 167368. doi :10.1016/j.jmb.2021.167368. PMID 34808226.
^ Moda F (2017). "Amplificación cíclica de priones infecciosos por plegamiento incorrecto de proteínas". Progreso en biología molecular y ciencia traslacional . 150 : 361–374. doi :10.1016/bs.pmbts.2017.06.016. ISBN.978-0-12-811226-7. Número de identificación personal 28838669.
^ Groschup MH, Kretzschmar HA, eds. (2001). Diagnóstico y patogenia de las enfermedades priónicas . Archivos de Virología. vol. 16. Nueva York: Springer. ISBN978-3-211-83530-2.
^ Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. (octubre de 1995). "La propagación de priones en ratones que expresan transgenes PrP humanos y quiméricos implica la interacción de la PrP celular con otra proteína". Cell . 83 (1): 79–90. doi : 10.1016/0092-8674(95)90236-8 . PMID 7553876. S2CID 15235574.
^ Johnson CJ, Pedersen JA, Chappell RJ, McKenzie D, Aiken JM (julio de 2007). "La transmisibilidad oral de la enfermedad priónica se ve reforzada por la unión a partículas del suelo". PLOS Pathogens . 3 (7): e93. doi : 10.1371/journal.ppat.0030093 . PMC 1904474 . PMID 17616973.
^ Tamgüney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, Palmer C, et al. (septiembre de 2009). "Los ciervos asintomáticos excretan priones infecciosos en las heces". Nature . 461 (7263): 529–532. Bibcode :2009Natur.461..529T. doi :10.1038/nature08289. PMC 3186440 . PMID 19741608.
^ Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, et al. (enero de 2011). "Los aerosoles transmiten priones a ratones inmunocompetentes e inmunodeficientes". PLOS Pathogens . 7 (1): e1001257. doi : 10.1371/journal.ppat.1001257 . PMC 3020930 . PMID 21249178.(Retractado, véase doi :10.1371/journal.ppat.1012396, PMID 39024193 . Si se trata de una cita intencional de un artículo retractado, reemplácelo con . ){{retracted|...}}{{retracted|...|intentional=yes}} Resumen para legos: Mackenzie D (13 de enero de 2011). "La enfermedad priónica puede propagarse a través del aire" . New Scientist .
^ Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, et al. (julio de 2024). "Retracción: los aerosoles transmiten priones a ratones inmunocompetentes e inmunodeficientes". PLOS Pathogens . 20 (7): e1012396. doi : 10.1371/journal.ppat.1012396 . PMC 11257221 . PMID 39024193.
^ Van Dorsselaer A, Carapito C, Delalande F, Schaeffer-Reiss C, Thierse D, Diemer H, et al. (marzo de 2011). "Detección de proteína priónica en productos de fertilidad inyectables derivados de la orina mediante un enfoque proteómico dirigido". PLOS ONE . 6 (3): e17815. Bibcode :2011PLoSO...617815V. doi : 10.1371/journal.pone.0017815 . PMC 3063168 . PMID 21448279.
^ Beecher C (1 de junio de 2015). «Un descubrimiento sorprendente sobre la enfermedad del desgaste crónico». Food Safety News . Archivado desde el original el 28 de abril de 2016. Consultado el 8 de abril de 2016 .
^ Pritzkow S, Morales R, Moda F, Khan U, Telling GC, Hoover E, et al. (mayo de 2015). "Las plantas herbáceas se unen, retienen, absorben y transportan priones infecciosos". Cell Reports . 11 (8): 1168–1175. doi :10.1016/j.celrep.2015.04.036. PMC 4449294 . PMID 25981035.
^ Qin K, O'Donnell M, Zhao RY (agosto de 2006). "Doppel: más rival que el doble del prión". Neurociencia . 141 (1): 1–8. doi :10.1016/j.neuroscience.2006.04.057. PMID 16781817. S2CID 28822120.
^ Race RE, Raymond GJ (febrero de 2004). "Inactivación de agentes de encefalopatía espongiforme transmisible (priones) por LpH ambiental". Journal of Virology . 78 (4): 2164–2165. doi :10.1128/JVI.78.4.2164-2165.2004. PMC 369477 . PMID 14747583.
^ Sutton JM, Dickinson J, Walker JT, Raven ND (septiembre de 2006). "Métodos para minimizar los riesgos de transmisión de la enfermedad de Creutzfeldt-Jakob mediante procedimientos quirúrgicos: ¿dónde establecer el estándar?". Enfermedades infecciosas clínicas . 43 (6): 757–764. doi : 10.1086/507030 . PMID 16912952.
^ Collins SJ, Lawson VA, Masters CL (enero de 2004). "Encefalopatías espongiformes transmisibles". Lancet . 363 (9402): 51–61. doi :10.1016/S0140-6736(03)15171-9. PMID 14723996. S2CID 23212525.
^ Brown P, Rau EH, Johnson BK, Bacote AE, Gibbs CJ, Gajdusek DC (marzo de 2000). "Nuevos estudios sobre la resistencia al calor del agente del scrapie adaptado al hámster: el umbral de supervivencia después de la incineración a 600 grados C sugiere una plantilla inorgánica de replicación". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 97 (7): 3418–3421. Bibcode :2000PNAS...97.3418B. doi : 10.1073/pnas.050566797 . PMC 16254 . PMID 10716712.
^ "Esterilización con ozono". Agencia de Protección de la Salud del Reino Unido. 14 de abril de 2005. Archivado desde el original el 10 de febrero de 2007. Consultado el 28 de febrero de 2010 .
^ Botsios S, Tittman S, Manuelidis L (2015). "La rápida descontaminación química de partículas infecciosas de ECJ y scrapie es paralela a los tratamientos que se sabe que alteran los microbios y las biopelículas". Virulence . 6 (8): 787–801. doi :10.1080/21505594.2015.1098804. PMC 4826107 . PMID 26556670.
^ Koga Y, Tanaka S, Sakudo A, Tobiume M, Aranishi M, Hirata A, et al. (marzo de 2014). "Proteólisis de proteína priónica anormal con una proteasa termoestable de Thermococcus kodakarensis KOD1". Applied Microbiology and Biotechnology . 98 (5): 2113–2120. doi :10.1007/s00253-013-5091-7. PMID 23880875. S2CID 2677641.
^ Eraña H, Pérez-Castro MÁ, García-Martínez S, Charco JM, López-Moreno R, Díaz-Dominguez CM, et al. (2020). "Un método novedoso, confiable y muy versátil para evaluar diferentes procedimientos de descontaminación de priones". Fronteras en Bioingeniería y Biotecnología . 8 : 589182. doi : 10.3389/fbioe.2020.589182 . PMC 7658626 . PMID 33195153.
^ Weissmann C, Enari M, Klöhn PC, Rossi D, Flechsig E (diciembre de 2002). "Transmisión de priones". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 99 (s 4): 16378–16383. Bibcode :2002PNAS...9916378W. doi : 10.1073/pnas.172403799 . PMC 139897 . PMID 12181490.
^ Zabel M, Ortega A (septiembre de 2017). "La ecología de los priones". Microbiology and Molecular Biology Reviews . 81 (3). doi :10.1128/MMBR.00001-17. PMC 5584314 . PMID 28566466.
^ Kuznetsova A, Cullingham C, McKenzie D, Aiken JM (noviembre de 2018). "Los ácidos húmicos del suelo degradan los priones de CWD y reducen la infectividad". PLOS Pathogens . 14 (11): e1007414. doi : 10.1371/journal.ppat.1007414 . PMC 6264147 . PMID 30496301.
^ Yuan Q, Eckland T, Telling G, Bartz J, Bartelt-Hunt S (febrero de 2015). "Mitigación de la infectividad de los priones y la capacidad de conversión mediante un proceso natural simulado: ciclos repetidos de secado y humectación". PLOS Pathogens . 11 (2): e1004638. doi : 10.1371/journal.ppat.1004638 . PMC 4335458 . PMID 25665187.
^ ab López-Pérez Ó, Badiola JJ, Bolea R, Ferrer I, Llorens F, Martín-Burriel I (27 de agosto de 2020). "Una actualización sobre la autofagia en enfermedades priónicas". Frontiers in Bioengineering and Biotechnology . 8 : 975. doi : 10.3389/fbioe.2020.00975 . PMC 7481332 . PMID 32984276.
^ Langeveld JP, Wang JJ, Van de Wiel DF, Shih GC, Garssen GJ, Bossers A, et al. (diciembre de 2003). "Degradación enzimática de la proteína priónica en el tronco encefálico de ganado vacuno y ovino infectados". The Journal of Infectious Diseases . 188 (11): 1782–1789. doi :10.1086/379664. PMID 14639552.
^ Okoroma EA, Purchase D, Garelick H, Morris R, Neale MH, Windl O, et al. (16 de julio de 2013). "Formulación enzimática capaz de degradar el prión de scrapie en condiciones de digestión suaves". PLOS ONE . 8 (7): e68099. Bibcode :2013PLoSO...868099O. doi : 10.1371/journal.pone.0068099 . PMC 3712960 . PMID 23874511.
^ Hui Z, Doi H, Kanouchi H, Matsuura Y, Mohri S, Nonomura Y, et al. (agosto de 2004). "La serina proteasa alcalina producida por Streptomyces sp. degrada PrP(Sc)". Comunicaciones de investigación bioquímica y biofísica . 321 (1): 45–50. doi :10.1016/j.bbrc.2004.06.100. PMID 15358213.
^ Snajder M, Vilfan T, Cernilec M, Rupreht R, Popović M, Juntes P, et al. (2012). "Degradación enzimática de PrPSc por una proteasa secretada de Aeropyrum pernix K1". PLOS ONE . 7 (6): e39548. Bibcode :2012PLoSO...739548S. doi : 10.1371/journal.pone.0039548 . PMC 3386259 . PMID 22761822.
^ Mitsuiki S, Hui Z, Matsumoto D, Sakai M, Moriyama Y, Furukawa K, et al. (mayo de 2006). "Degradación de PrP(Sc) por la proteasa queratinolítica de Nocardiopsis sp. TOA-1". Biociencia, biotecnología y bioquímica . 70 (5): 1246–1248. doi :10.1271/bbb.70.1246. PMID 16717429.
^ Hsu RL, Lee KT, Wang JH, Lee LY, Chen RP (enero de 2009). "Capacidad de degradación de amiloide de la nattoquinasa de Bacillus subtilis natto". Journal of Agricultural and Food Chemistry . 57 (2): 503–508. doi :10.1021/jf803072r. PMID 19117402.
^ Booth CJ, Johnson CJ, Pedersen JA (abril de 2013). "Inactivación microbiana y enzimática de priones en ambientes de suelo". Soil Biology and Biochemistry . 59 : 1–15. Bibcode :2013SBiBi..59....1B. doi :10.1016/j.soilbio.2012.12.016. ISSN 0038-0717.
^ Dickinson J, Murdoch H, Dennis MJ, Hall GA, Bott R, Crabb WD, et al. (mayo de 2009). "Descontaminación de la proteína priónica (BSE301V) utilizando una proteasa modificada genéticamente". The Journal of Hospital Infection . 72 (1): 65–70. doi :10.1016/j.jhin.2008.12.007. PMID 19201054.
^ Johnson CJ, Bennett JP, Biro SM, Duque-Velasquez JC, Rodriguez CM, Bessen RA, et al. (mayo de 2011). "Degradación de la proteína priónica asociada a la enfermedad por una serina proteasa de líquenes". PLOS ONE . 6 (5): e19836. Bibcode :2011PLoSO...619836J. doi : 10.1371/journal.pone.0019836 . PMC 3092769 . PMID 21589935.
^ Lindquist S, Krobitsch S, Li L, Sondheimer N (febrero de 2001). "Investigación de la herencia basada en la conformación de proteínas y la enfermedad en levaduras". Philosophical Transactions of the Royal Society of London. Serie B, Biological Sciences . 356 (1406): 169–176. doi :10.1098/rstb.2000.0762. PMC 1088422 . PMID 11260797.
^ Aguzzi A (enero de 2008). "Descifrando las cepas priónicas con biología celular y química orgánica". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 105 (1): 11–12. Bibcode :2008PNAS..105...11A. doi : 10.1073/pnas.0710824105 . PMC 2224168 . PMID 18172195.
^ Dong J, Bloom JD, Goncharov V, Chattopadhyay M, Millhauser GL, Lynn DG, et al. (noviembre de 2007). "Investigación del papel de las repeticiones de PrP en la conversión conformacional y el ensamblaje amiloide de priones de levadura quiméricos". The Journal of Biological Chemistry . 282 (47): 34204–34212. doi : 10.1074/jbc.M704952200 . PMC 2262835 . PMID 17893150.
^ Newby GA, Lindquist S (junio de 2013). "Bendiciones disfrazadas: beneficios biológicos de los mecanismos priónicos". Tendencias en biología celular . 23 (6): 251–259. doi :10.1016/j.tcb.2013.01.007. hdl : 1721.1/103966 . PMID 23485338.
^ Halfmann R, Lindquist S (octubre de 2010). "Epigenética en extremo: priones y herencia de rasgos adquiridos ambientalmente". Science . 330 (6004): 629–632. Bibcode :2010Sci...330..629H. doi :10.1126/science.1191081. PMID 21030648. S2CID 206527151.
^ Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S (febrero de 2012). "Los priones son un mecanismo común de herencia fenotípica en levaduras silvestres". Nature . 482 (7385): 363–368. Bibcode :2012Natur.482..363H. doi :10.1038/nature10875. PMC 3319070 . PMID 22337056.
^ Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, et al. (junio de 2010). "El determinante no mendeliano [ISP+] en la levadura es una forma priónica que reside en el núcleo del regulador transcripcional global Sfp1". Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 107 (23): 10573–10577. Bibcode :2010PNAS..10710573R. doi : 10.1073/pnas.1005949107 . PMC 2890785 . PMID 20498075.
^ ab Aguzzi A, Lakkaraju AK, Frontzek K (enero de 2018). "Hacia la terapia de las enfermedades priónicas humanas" (PDF) . Revisión anual de farmacología y toxicología . 58 (1): 331–351. doi :10.1146/annurev-pharmtox-010617-052745. PMID 28961066. Archivado (PDF) del original el 12 de marzo de 2020. Consultado el 5 de marzo de 2020 .
^ "Prion Clinic – Tratamientos farmacológicos". 13 de septiembre de 2017. Archivado desde el original el 29 de enero de 2020. Consultado el 29 de enero de 2020 .
^ abc King OD, Gitler AD, Shorter J (junio de 2012). "La punta del iceberg: proteínas de unión al ARN con dominios similares a priones en enfermedades neurodegenerativas". Brain Research . 1462 : 61–80. doi :10.1016/j.brainres.2012.01.016. PMC 3372647 . PMID 22445064.
^ Goedert M (agosto de 2015). "NEURODEGENERACIÓN. Enfermedades de Alzheimer y Parkinson: el concepto de prión en relación con Aβ, tau y α-sinucleína ensamblados". Science . 349 (6248): 1255555. doi :10.1126/science.1255555. PMID 26250687. S2CID 206558562.
^ Murakami T, Ishiguro N, Higuchi K (marzo de 2014). "Transmisión de amiloidosis AA sistémica en animales". Patología veterinaria . 51 (2): 363–371. doi : 10.1177/0300985813511128 . PMID 24280941.
^ Jucker M, Walker LC (septiembre de 2013). "Autopropagación de agregados proteicos patógenos en enfermedades neurodegenerativas". Nature . 501 (7465): 45–51. Bibcode :2013Natur.501...45J. doi :10.1038/nature12481. PMC 3963807 . PMID 24005412.
^ Alberti S, Halfmann R, King O, Kapila A, Lindquist S (abril de 2009). "Un estudio sistemático identifica priones y arroja luz sobre las características de secuencia de las proteínas prionogénicas". Cell . 137 (1): 146–158. doi :10.1016/j.cell.2009.02.044. PMC 2683788 . PMID 19345193.
^ ab Eisenberg D, Jucker M (marzo de 2012). "El estado amiloide de las proteínas en enfermedades humanas". Cell . 148 (6): 1188–1203. doi :10.1016/j.cell.2012.02.022. PMC 3353745 . PMID 22424229.
^ Ayers JI, Prusiner SB (abril de 2020). "Proteína priónica: mediadora de toxicidad en múltiples proteinopatías". Nature Reviews. Neurología . 16 (4): 187–188. doi :10.1038/s41582-020-0332-8. PMID 32123368. S2CID 211728879.
^ Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, et al. (marzo de 2013). "Las mutaciones en dominios similares a priones en hnRNPA2B1 y hnRNPA1 causan proteinopatía multisistémica y ELA". Nature . 495 (7442): 467–473. Bibcode :2013Natur.495..467K. doi :10.1038/nature11922. PMC 3756911 . PMID 23455423.
^ "¿Qué son las armas biológicas?". Naciones Unidas, Oficina de Asuntos de Desarme. Archivado desde el original el 21 de mayo de 2021. Consultado el 21 de mayo de 2021 .
^ "Priones: el peligro de las armas bioquímicas" (PDF) . Archivado (PDF) del original el 9 de diciembre de 2020 . Consultado el 21 de mayo de 2021 .
^ "La próxima plaga: los priones son diminutos, misteriosos y aterradores". American Council on Science and Health. 20 de marzo de 2017. Archivado desde el original el 21 de mayo de 2021. Consultado el 21 de mayo de 2021 .
^ "¿Priones como armas biológicas? Mucho ruido y pocas nueces; o preocupaciones acertadas sobre las pequeñas proteínas utilizadas en la guerra biológica". Defense iQ. 13 de septiembre de 2019. Archivado desde el original el 21 de mayo de 2021. Consultado el 21 de mayo de 2021 .
^ "Cómo surgieron los priones: una breve historia - Enfermedades infecciosas: superbacterias, ciencia y sociedad". Archivado desde el original el 17 de septiembre de 2021 . Consultado el 17 de septiembre de 2021 .
^ Ness A, Aiken J, McKenzie D (diciembre de 2023). "Tembladera ovina y rabia de ciervo en Inglaterra antes de 1800". Prion . 17 (1): 7–15. doi :10.1080/19336896.2023.2166749. PMC 9858414 . PMID 36654484.
^ Alper T, Cramp WA, Haig DA, Clarke MC (mayo de 1967). "¿Se replica el agente del scrapie sin ácido nucleico?". Nature . 214 (5090): 764–766. Bibcode :1967Natur.214..764A. doi :10.1038/214764a0. PMID 4963878. S2CID 4195902.
^ Griffith JS (septiembre de 1967). "Autorreplicación y scrapie". Nature . 215 (5105): 1043–1044. Código Bibliográfico :1967Natur.215.1043G. doi :10.1038/2151043a0. PMID 4964084. S2CID 4171947.
^ Field EJ (septiembre de 1966). "Experimentos de transmisión con esclerosis múltiple: un informe provisional". British Medical Journal . 2 (5513): 564–565. doi :10.1136/bmj.2.5513.564. PMC 1943767 . PMID 5950508.
^ Adams DH, Field EJ (septiembre de 1968). "El proceso infeccioso en el scrapie". Lancet . 2 (7570): 714–716. doi :10.1016/s0140-6736(68)90754-x. PMID 4175093.
^ Field EJ, Farmer F, Caspary EA, Joyce G (abril de 1969). "Susceptibilidad del agente causante de la tembladera a la radiación ionizante". Nature . 5188. 222 (5188): 90–91. Bibcode :1969Natur.222...90F. doi :10.1038/222090a0. PMID 4975649. S2CID 4195610.
^ Griffith JS (septiembre de 1967). "Autorreplicación y scrapie". Nature . 215 (5105): 1043–1044. Código Bibliográfico :1967Natur.215.1043G. doi :10.1038/2151043a0. PMID 4964084. S2CID 4171947.
^ ab Bolton D (1 de enero de 2004). "Priones, la hipótesis de las proteínas y revoluciones científicas". En Nunnally BK, Krull IS (eds.). Priones y enfermedad de las vacas locas . Marcel Dekker. págs. 21–60. ISBN978-0-203-91297-3. Archivado del original el 22 de marzo de 2022 . Consultado el 27 de julio de 2018 – vía ResearchGate.
^ Crick F (agosto de 1970). "Dogma central de la biología molecular". Nature . 227 (5258): 561–563. Bibcode :1970Natur.227..561C. doi :10.1038/227561a0. PMID 4913914. S2CID 4164029.
^ Coffin JM, Fan H (septiembre de 2016). "El descubrimiento de la transcriptasa inversa". Revisión anual de virología . 3 (1): 29–51. doi : 10.1146/annurev-virology-110615-035556 . PMID 27482900.
^ Taubes G (diciembre de 1986). "El juego del nombre es fama. Pero ¿es ciencia?". Discover . 7 (12): 28–41.
^ "El Premio Nobel de Fisiología o Medicina, 1997". NobelPrize.org. Archivado desde el original el 9 de agosto de 2018. Consultado el 28 de febrero de 2010. El Premio Nobel de Fisiología o Medicina de 1997 fue otorgado a Stanley B. Prusiner "por su descubrimiento de los priones, un nuevo principio biológico de infección".
^ Frazer J. "Los priones son para siempre". Red de blogs de Scientific American . Archivado desde el original el 4 de enero de 2022. Consultado el 28 de diciembre de 2021 .



{kind=link}