.jpg/1280px-Clinical_Trial_Participant_Receives_Injection_(34033294061).jpg)
Los ensayos clínicos son estudios de investigación biomédica o conductual prospectivos en participantes humanos diseñados para responder preguntas específicas sobre intervenciones biomédicas o conductuales, incluidos nuevos tratamientos (como nuevas vacunas , medicamentos , opciones dietéticas , suplementos dietéticos y dispositivos médicos ) e intervenciones conocidas que justifican un estudio y una comparación más profundos. Los ensayos clínicos generan datos sobre la dosis, la seguridad y la eficacia. [1] [2] Se llevan a cabo solo después de haber recibido la aprobación de la autoridad sanitaria/comité de ética en el país donde se busca la aprobación de la terapia. Estas autoridades son responsables de examinar la relación riesgo/beneficio del ensayo: su aprobación no significa que la terapia sea "segura" o efectiva, solo que el ensayo puede llevarse a cabo.
Dependiendo del tipo de producto y la etapa de desarrollo, los investigadores inicialmente inscriben voluntarios o pacientes en pequeños estudios piloto y luego realizan estudios comparativos a mayor escala. Los ensayos clínicos pueden variar en tamaño y costo, y pueden involucrar un solo centro de investigación o múltiples centros , en un país o en varios países. El diseño del estudio clínico tiene como objetivo garantizar la validez científica y la reproducibilidad de los resultados.
Los costos de los ensayos clínicos pueden ascender a miles de millones de dólares por cada fármaco aprobado [3], y el proceso completo de ensayo hasta su aprobación puede requerir de 7 a 15 años [4] [5] El patrocinador puede ser una organización gubernamental o una empresa farmacéutica , de biotecnología o de dispositivos médicos. Ciertas funciones necesarias para el ensayo, como el seguimiento y el trabajo de laboratorio, pueden ser gestionadas por un socio externo, como una organización de investigación por contrato o un laboratorio central. Sólo el 10 por ciento de todos los fármacos que se inician en ensayos clínicos en humanos se convierten en fármacos aprobados [6] .
Algunos ensayos clínicos involucran sujetos sanos sin condiciones médicas preexistentes . Otros ensayos clínicos se realizan con personas con condiciones de salud específicas que están dispuestas a probar un tratamiento experimental. Los experimentos piloto se llevan a cabo para obtener información para el diseño del ensayo clínico posterior. [ cita requerida ]
Los ensayos de tratamientos médicos tienen dos objetivos: saber si funcionan lo suficientemente bien, lo que se denomina "eficacia" o "efectividad"; y saber si son lo suficientemente seguros, lo que se denomina "seguridad". [1] Ninguno de estos criterios es absoluto; tanto la seguridad como la eficacia se evalúan en relación con el uso previsto del tratamiento, los otros tratamientos disponibles y la gravedad de la enfermedad o afección. Los beneficios deben ser mayores que los riesgos. [7] [8] : 8 Por ejemplo, muchos medicamentos para tratar el cáncer tienen efectos secundarios graves que no serían aceptables para un analgésico de venta libre, pero los medicamentos contra el cáncer han sido aprobados porque se utilizan bajo el cuidado de un médico y se utilizan para una afección potencialmente mortal. [9]
En los EE. UU., los ancianos constituyen el 14% de la población, mientras que consumen más de un tercio de los medicamentos. [10] Las personas mayores de 55 años (o una edad límite similar) a menudo son excluidas de los ensayos porque sus mayores problemas de salud y el consumo de medicamentos complican la interpretación de los datos, y porque tienen una capacidad fisiológica diferente a la de las personas más jóvenes. Los niños y las personas con afecciones médicas no relacionadas también son excluidos con frecuencia. [11] Las mujeres embarazadas a menudo son excluidas debido a los posibles riesgos para el feto .
El patrocinador diseña el ensayo en coordinación con un panel de investigadores clínicos expertos, lo que incluye qué tratamientos alternativos o existentes se compararán con el nuevo fármaco y qué tipo(s) de pacientes podrían beneficiarse. Si el patrocinador no puede obtener suficientes sujetos de prueba en un lugar, se reclutan investigadores de otros lugares para que se unan al estudio. [ cita requerida ]
Durante el ensayo, los investigadores reclutan sujetos con las características predeterminadas, administran el tratamiento o los tratamientos y recopilan datos sobre la salud de los sujetos durante un período de tiempo definido. Los datos incluyen mediciones como los signos vitales , la concentración del fármaco en estudio en la sangre o los tejidos, los cambios en los síntomas y si se produce una mejora o un empeoramiento de la afección a la que se dirige el fármaco en estudio. Los investigadores envían los datos al patrocinador del ensayo, quien luego analiza los datos agrupados mediante pruebas estadísticas . [ cita requerida ]
Algunos ejemplos de objetivos de ensayos clínicos incluyen evaluar la seguridad y la eficacia relativa de un medicamento o dispositivo: [ cita requerida ]
Si bien la mayoría de los ensayos clínicos prueban una alternativa a la nueva intervención, algunos se amplían a tres o cuatro y pueden incluir un placebo . [ cita requerida ]
A excepción de los ensayos pequeños realizados en un solo lugar, el diseño y los objetivos se especifican en un documento llamado protocolo de ensayo clínico . El protocolo es el "manual de operaciones" del ensayo y garantiza que todos los investigadores realicen el ensayo de la misma manera en sujetos similares y que los datos sean comparables entre todos los sujetos. [ cita requerida ]
Como un ensayo está diseñado para probar hipótesis y monitorear y evaluar rigurosamente los resultados, puede verse como una aplicación del método científico , específicamente la etapa experimental. [ cita requerida ]
Los ensayos clínicos más comunes evalúan nuevos productos farmacéuticos, dispositivos médicos, productos biológicos , ensayos de diagnóstico , terapias psicológicas u otras intervenciones. [12] Es posible que se requieran ensayos clínicos antes de que una autoridad reguladora nacional [13] apruebe la comercialización de la innovación.
De manera similar a los medicamentos, los fabricantes de dispositivos médicos en los Estados Unidos deben realizar ensayos clínicos para obtener la aprobación previa a la comercialización . [14] Los ensayos de dispositivos pueden comparar un dispositivo nuevo con una terapia establecida, o pueden comparar dispositivos similares entre sí. Un ejemplo de lo primero en el campo de la cirugía vascular es el ensayo Open versus Endovascular Repair (OVER) para el tratamiento del aneurisma aórtico abdominal , que comparó la antigua técnica de reparación aórtica abierta con el nuevo dispositivo de reparación endovascular del aneurisma . [15] Un ejemplo de lo último son los ensayos clínicos sobre dispositivos mecánicos utilizados en el tratamiento de la incontinencia urinaria femenina adulta . [16]
De manera similar a los medicamentos, los procedimientos médicos o quirúrgicos pueden estar sujetos a ensayos clínicos, [17] como la comparación de diferentes enfoques quirúrgicos en el tratamiento de los fibromas por subfertilidad . [18] Sin embargo, cuando los ensayos clínicos son poco éticos o logísticamente imposibles en el ámbito quirúrgico, se reemplazarán los estudios de casos y controles . [19]
Además de participar en un ensayo clínico, los miembros del público pueden colaborar activamente con los investigadores en el diseño y la realización de investigaciones clínicas . Esto se conoce como participación del paciente y del público (PPI). La participación del público implica una asociación de trabajo entre pacientes, cuidadores, personas con experiencia vivida e investigadores para dar forma e influir en lo que hace el investigador y cómo lo hace. [20] La PPI puede mejorar la calidad de la investigación y hacerla más relevante y accesible. Las personas con experiencia actual o pasada de enfermedad pueden proporcionar una perspectiva diferente a la de los profesionales y complementar su conocimiento. A través de su conocimiento personal, pueden identificar temas de investigación que son relevantes e importantes para quienes viven con una enfermedad o utilizan un servicio. También pueden ayudar a que la investigación se base más en las necesidades de las comunidades específicas de las que forman parte. Los contribuyentes públicos también pueden garantizar que la investigación se presente en un lenguaje sencillo que sea claro para la sociedad en general y los grupos específicos para los que es más relevante. [21]

Aunque los primeros experimentos médicos se realizaron con frecuencia, en general no se utilizó un grupo de control para proporcionar una comparación precisa para demostrar la eficacia de la intervención. Por ejemplo, Lady Mary Wortley Montagu , que hizo campaña por la introducción de la inoculación (en aquel entonces llamada variolación) para prevenir la viruela , hizo que siete prisioneros que habían sido condenados a muerte se sometieran a la variolación a cambio de su vida. Aunque sobrevivieron y no contrajeron la viruela, no hubo un grupo de control para evaluar si este resultado se debió a la inoculación o a algún otro factor. Experimentos similares realizados por Edward Jenner sobre su vacuna contra la viruela fueron igualmente defectuosos desde el punto de vista conceptual. [22]
El primer ensayo clínico adecuado fue realizado por el médico escocés James Lind . [23] La enfermedad del escorbuto , ahora conocida por ser causada por una deficiencia de vitamina C , a menudo tenía efectos terribles en el bienestar de la tripulación de los viajes oceánicos de larga distancia. En 1740, el catastrófico resultado de la circunnavegación de Anson atrajo mucha atención en Europa; de 1900 hombres, 1400 habían muerto, la mayoría de ellos supuestamente por haber contraído escorbuto. [24] John Woodall , un cirujano militar inglés de la Compañía Británica de las Indias Orientales , había recomendado el consumo de frutas cítricas desde el siglo XVII, pero su uso no se generalizó. [25]
Lind realizó el primer ensayo clínico sistemático en 1747. [26] Incluyó un suplemento dietético de calidad ácida en el experimento después de dos meses en el mar, cuando el barco ya estaba afligido por el escorbuto. Dividió a doce marineros escorbúticos en seis grupos de dos. Todos recibieron la misma dieta pero, además, al grupo uno se le dio un cuarto de galón de sidra diariamente, al grupo dos veinticinco gotas de elixir de vitriolo ( ácido sulfúrico ), al grupo tres seis cucharadas de vinagre , al grupo cuatro media pinta de agua de mar, al grupo cinco recibió dos naranjas y un limón , y al último grupo una pasta picante más un trago de agua de cebada . El tratamiento del grupo cinco se detuvo después de seis días cuando se quedaron sin fruta, pero para entonces un marinero estaba en condiciones de cumplir con el deber mientras que el otro casi se había recuperado. Aparte de eso, solo el grupo uno también mostró algún efecto de su tratamiento. [27] Cada año, el 20 de mayo se celebra el Día de los Ensayos Clínicos en honor a la investigación de Lind. [28]
Después de 1750, la disciplina comenzó a tomar su forma moderna. [29] [30] El médico inglés John Haygarth demostró la importancia de un grupo de control para la correcta identificación del efecto placebo en su célebre estudio del remedio ineficaz llamado tractores de Perkin . El eminente médico Sir William Gull, primer baronet, realizó más trabajos en esa dirección en la década de 1860. [22]
Frederick Akbar Mahomed (fallecido en 1884), que trabajaba en el Guy's Hospital de Londres , hizo importantes contribuciones al proceso de ensayos clínicos, donde "separó la nefritis crónica con hipertensión secundaria de lo que ahora llamamos hipertensión esencial . También fundó el Registro de Investigación Colectiva para la Asociación Médica Británica ; esta organización recopiló datos de médicos que ejercían fuera del ámbito hospitalario y fue la precursora de los ensayos clínicos colaborativos modernos". [31]

Las ideas de Sir Ronald A. Fisher todavía juegan un papel en los ensayos clínicos. Mientras trabajaba para la estación experimental de Rothamsted en el campo de la agricultura, Fisher desarrolló sus Principios de diseño experimental en la década de 1920 como una metodología precisa para el diseño adecuado de experimentos. Entre sus ideas principales se incluyen la importancia de la aleatorización : la asignación aleatoria de individuos a diferentes grupos para el experimento; [32] la replicación : para reducir la incertidumbre , las mediciones deben repetirse y los experimentos replicarse para identificar fuentes de variación; [33] el bloqueo : para organizar las unidades experimentales en grupos de unidades que sean similares entre sí, y así reducir las fuentes irrelevantes de variación; el uso de experimentos factoriales : eficientes para evaluar los efectos y las posibles interacciones de varios factores independientes. [22] De estos, el diseño factorial y de bloqueo rara vez se aplican en ensayos clínicos, porque las unidades experimentales son sujetos humanos y normalmente solo hay una intervención independiente: el tratamiento. [ cita requerida ]
El Consejo Británico de Investigación Médica reconoció oficialmente la importancia de los ensayos clínicos a partir de la década de 1930. El consejo creó el Comité de Ensayos Terapéuticos para asesorar y ayudar en la organización de ensayos clínicos adecuadamente controlados sobre nuevos productos que parezcan tener valor en el tratamiento de enfermedades desde el punto de vista experimental. [22]
El primer ensayo curativo aleatorizado fue llevado a cabo en la Unidad de Investigación de Tuberculosis del MRC por Sir Geoffrey Marshall (1887-1982). El ensayo, llevado a cabo entre 1946 y 1947, tuvo como objetivo probar la eficacia de la estreptomicina química para curar la tuberculosis pulmonar . El ensayo fue doble ciego y controlado con placebo . [34]
La metodología de los ensayos clínicos fue desarrollada por Sir Austin Bradford Hill , que había participado en los ensayos de estreptomicina. A partir de la década de 1920, Hill aplicó la estadística a la medicina, asistiendo a las conferencias del renombrado matemático Karl Pearson , entre otros. Se hizo famoso por un estudio histórico realizado en colaboración con Richard Doll sobre la correlación entre el tabaquismo y el cáncer de pulmón . Llevaron a cabo un estudio de casos y controles en 1950, que comparó a pacientes de cáncer de pulmón con un control emparejado y también comenzaron un estudio prospectivo sostenido a largo plazo sobre la cuestión más amplia del tabaquismo y la salud, que implicó estudiar los hábitos de tabaquismo y la salud de más de 30.000 médicos durante un período de varios años. Su certificado de elección para la Royal Society lo llamó "... el líder en el desarrollo en medicina de los métodos experimentales precisos que ahora se utilizan a nivel nacional e internacional en la evaluación de nuevos agentes terapéuticos y profilácticos ".
El 20 de mayo se celebra el Día Internacional de los Ensayos Clínicos. [35]
Las siglas utilizadas en los títulos de los ensayos clínicos suelen ser artificiales y han sido objeto de burla. [36]
Los ensayos clínicos se clasifican según el objetivo de investigación creado por los investigadores. [12]
Los ensayos se clasifican según su finalidad. Una vez que se concede la aprobación para la investigación en seres humanos al patrocinador del ensayo, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) organiza y supervisa los resultados de los ensayos según el tipo: [12]
Los ensayos clínicos se llevan a cabo normalmente en cuatro fases, y cada fase utiliza un número diferente de sujetos y tiene un propósito diferente para centrarse en la identificación de un efecto específico. [12]
Los ensayos clínicos de nuevos medicamentos se clasifican comúnmente en cinco fases. Cada fase del proceso de aprobación de un medicamento se considera un ensayo clínico independiente. El proceso de desarrollo de un medicamento normalmente se desarrolla a través de las fases I a IV durante muchos años, con frecuencia durante una década o más. Si el medicamento pasa con éxito las fases I, II y III, generalmente será aprobado por la autoridad reguladora nacional para su uso en la población general. [12] Los ensayos de fase IV se realizan después de que el medicamento, diagnóstico o dispositivo recientemente aprobado se comercializa, y brindan una evaluación sobre los riesgos, los beneficios o los mejores usos. [12]
Una distinción fundamental en la práctica basada en la evidencia es entre estudios observacionales y ensayos controlados aleatorios . [45] Los tipos de estudios observacionales en epidemiología , como el estudio de cohorte y el estudio de casos y controles , proporcionan evidencia menos convincente que el ensayo controlado aleatorio. [45] En los estudios observacionales, los investigadores evalúan retrospectivamente las asociaciones entre los tratamientos administrados a los participantes y su estado de salud, con potencial para errores considerables en el diseño y la interpretación. [46]
Un ensayo controlado aleatorio puede proporcionar evidencia convincente de que el tratamiento en estudio causa un efecto en la salud humana. [45]
Algunos ensayos de fármacos de fase II y la mayoría de los de fase III están diseñados como aleatorizados, doble ciego y controlados con placebo . [ cita requerida ]
Los estudios clínicos que tienen un número pequeño de sujetos pueden ser "patrocinados" por investigadores individuales o un grupo pequeño de investigadores, y están diseñados para probar preguntas simples o la viabilidad de expandir la investigación para un ensayo controlado aleatorio más completo. [47]
Los estudios clínicos pueden ser "patrocinados" (financiados y organizados) por instituciones académicas, compañías farmacéuticas, entidades gubernamentales e incluso grupos privados. Se realizan ensayos para nuevos medicamentos, biotecnología, ensayos de diagnóstico o dispositivos médicos para determinar su seguridad y eficacia antes de ser presentados para la revisión regulatoria que determinaría la aprobación para su comercialización. [ cita requerida ]
En los casos en que administrar un placebo a una persona que padece una enfermedad puede resultar poco ético, se pueden realizar en su lugar ensayos de "comparación activa" (también conocidos como "control activo"). [48] En los ensayos con un grupo de control activo, los sujetos reciben el tratamiento experimental o un tratamiento previamente aprobado con eficacia conocida. En otros casos, los patrocinadores pueden realizar un ensayo de comparación activa para establecer una afirmación de eficacia relativa al comparador activo en lugar del placebo en el etiquetado . [ cita requerida ]
Un protocolo maestro incluye varios subestudios, que pueden tener diferentes objetivos e implicar esfuerzos coordinados para evaluar uno o más productos médicos en una o más enfermedades o afecciones dentro de la estructura general del estudio. Los ensayos que podrían desarrollar un protocolo maestro incluyen el ensayo paraguas (múltiples productos médicos para una sola enfermedad), el ensayo de plataforma (múltiples productos para una sola enfermedad que ingresan y salen de la plataforma) y el ensayo de canasta (un producto médico para múltiples enfermedades o subtipos de enfermedades). [49]
Las pruebas genéticas permiten a los investigadores agrupar a los pacientes según su perfil genético, administrar medicamentos basados en ese perfil a ese grupo y comparar los resultados. Pueden participar varias empresas, cada una de las cuales aporta un medicamento diferente. El primer enfoque de este tipo se centra en el cáncer de células escamosas , que incluye distintas alteraciones genéticas de un paciente a otro. Amgen, AstraZeneca y Pfizer participan, y es la primera vez que trabajan juntas en un ensayo de última etapa. Los pacientes cuyos perfiles genómicos no coinciden con ninguno de los medicamentos del ensayo reciben un medicamento diseñado para estimular el sistema inmunológico para que ataque el cáncer. [50]
Un protocolo de ensayo clínico es un documento que se utiliza para definir y gestionar el ensayo. Lo prepara un grupo de expertos. Se espera que todos los investigadores del estudio cumplan estrictamente con el protocolo. [ cita requerida ]
El protocolo describe la justificación científica, los objetivos, el diseño, la metodología, las consideraciones estadísticas y la organización del ensayo planificado. Los detalles del ensayo se proporcionan en los documentos a los que se hace referencia en el protocolo, como un folleto para el investigador . [ cita requerida ]
El protocolo contiene un plan de estudio preciso para garantizar la seguridad y la salud de los sujetos del ensayo y para proporcionar una plantilla exacta para la realización del ensayo por parte de los investigadores. Esto permite combinar los datos de todos los investigadores/centros. El protocolo también informa a los administradores del estudio (a menudo una organización de investigación por contrato ). [ cita requerida ]
El formato y el contenido de los protocolos de ensayos clínicos patrocinados por empresas farmacéuticas, biotecnológicas o de dispositivos médicos en los Estados Unidos, la Unión Europea o Japón se han estandarizado para seguir las pautas de Buenas Prácticas Clínicas [51] emitidas por la Conferencia Internacional de Armonización (ICH). [52] Las autoridades reguladoras de Canadá , China , Corea del Sur y el Reino Unido también siguen las pautas de la ICH. Revistas como Trials alientan a los investigadores a publicar sus protocolos.

Los ensayos clínicos reclutan a los sujetos del estudio para que firmen un documento que representa su " consentimiento informado ". [53] El documento incluye detalles como su propósito, duración, procedimientos requeridos, riesgos, beneficios potenciales, contactos clave y requisitos institucionales. [54] Luego, el participante decide si firma el documento. El documento no es un contrato, ya que el participante puede retirarse en cualquier momento sin penalización. [ cita requerida ]
El consentimiento informado es un proceso legal en el que se le informa al participante sobre hechos clave antes de decidir si participará o no. [53] Los investigadores explican los detalles del estudio en términos que el sujeto pueda comprender. La información se presenta en el idioma nativo del sujeto. Por lo general, los niños no pueden brindar su consentimiento informado de manera autónoma, pero, dependiendo de su edad y otros factores, se les puede exigir que brinden su asentimiento informado. [ cita requerida ]
En cualquier ensayo clínico, el número de sujetos, también llamado tamaño de la muestra, tiene un gran impacto en la capacidad de detectar y medir de manera confiable los efectos de la intervención. Esta capacidad se describe como su " poder ", que debe calcularse antes de iniciar un estudio para determinar si el estudio vale sus costos. [55] En general, un mayor tamaño de muestra aumenta el poder estadístico, también el costo.
El poder estadístico estima la capacidad de un ensayo para detectar una diferencia de un tamaño particular (o mayor) entre los grupos de tratamiento y control. Por ejemplo, un ensayo de un fármaco reductor de lípidos versus placebo con 100 pacientes en cada grupo podría tener un poder de 0,90 para detectar una diferencia entre los grupos de placebo y de ensayo que reciben una dosis de 10 mg/dl o más, pero solo 0,70 para detectar una diferencia de 6 mg/dl. [ cita requerida ]
La simple administración de un tratamiento puede tener efectos no específicos. Estos se controlan mediante la inclusión de pacientes que reciben únicamente un placebo. Los sujetos se asignan aleatoriamente sin informarles a qué grupo pertenecen. Muchos ensayos son doble ciego, de modo que los investigadores no saben a qué grupo está asignado un sujeto.
La asignación de un sujeto a un grupo placebo puede plantear un problema ético si viola su derecho a recibir el mejor tratamiento disponible. La Declaración de Helsinki ofrece directrices al respecto.
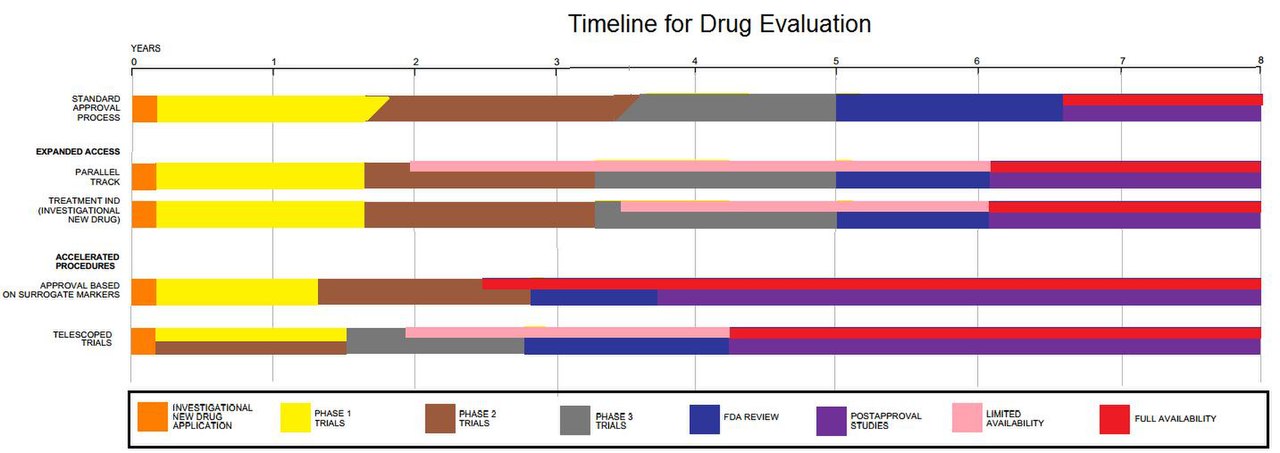
Los ensayos clínicos son sólo una pequeña parte de la investigación que se lleva a cabo para desarrollar un nuevo tratamiento. Por ejemplo, los medicamentos potenciales primero tienen que ser descubiertos, purificados, caracterizados y probados en laboratorios (en estudios con células y animales) antes de ser sometidos a ensayos clínicos. En total, se prueban alrededor de 1.000 medicamentos potenciales antes de que sólo uno llegue al punto de ser probado en un ensayo clínico. [56] Por ejemplo, un nuevo medicamento contra el cáncer tiene, en promedio, seis años de investigación detrás antes de que llegue a los ensayos clínicos. Pero el principal obstáculo para que los nuevos medicamentos contra el cáncer estén disponibles es el tiempo que lleva completar los ensayos clínicos. En promedio, pasan unos ocho años desde el momento en que un medicamento contra el cáncer entra en los ensayos clínicos hasta que recibe la aprobación de las agencias reguladoras para su venta al público. [57] Los medicamentos para otras enfermedades tienen plazos similares.
Algunas razones por las que un ensayo clínico podría durar varios años:
Un ensayo clínico también podría incluir un período de seguimiento posterior al estudio más extenso, que puede durar meses o años, para las personas que han participado en el ensayo, una denominada "fase de extensión", cuyo objetivo es identificar el impacto a largo plazo del tratamiento. [58]
El mayor obstáculo para completar los estudios es la escasez de participantes. Todos los ensayos de medicamentos y muchos de los dispositivos se dirigen a un subconjunto de la población, lo que significa que no todos pueden participar. Algunos ensayos de medicamentos requieren que los pacientes presenten combinaciones inusuales de características de la enfermedad. Es un desafío encontrar a los pacientes adecuados y obtener su consentimiento, especialmente cuando es posible que no reciban ningún beneficio directo (porque no se les paga, el medicamento en estudio aún no ha demostrado que funcione o el paciente puede recibir un placebo). En el caso de los pacientes con cáncer, menos del 5% de los adultos con cáncer participarán en ensayos de medicamentos. Según la Asociación de Fabricantes e Investigadores Farmacéuticos de Estados Unidos (PhRMA), en 2005 se estaban probando unos 400 medicamentos contra el cáncer en ensayos clínicos. No todos ellos resultarán útiles, pero los que lo sean pueden demorar su aprobación porque el número de participantes es muy bajo. [59]
En el caso de ensayos clínicos que impliquen posibles influencias estacionales (como alergias transmitidas por el aire , trastorno afectivo estacional , gripe y enfermedades de la piel ), el estudio puede realizarse durante una parte limitada del año (como la primavera para las alergias al polen), cuando se puede probar el medicamento. [60] [61]
Los ensayos clínicos que no involucran un medicamento nuevo suelen tener una duración mucho más corta (las excepciones son los estudios epidemiológicos, como el Nurses' Health Study ).
Los ensayos clínicos diseñados por un investigador local y (en los EE. UU.) los ensayos clínicos financiados por el gobierno federal casi siempre son administrados por el investigador que diseñó el estudio y solicitó la subvención. Los estudios de dispositivos a pequeña escala pueden ser administrados por la empresa patrocinadora. Los ensayos clínicos de nuevos medicamentos generalmente son administrados por una organización de investigación por contrato (CRO) contratada por la empresa patrocinadora. El patrocinador proporciona el medicamento y la supervisión médica. Se contrata a una CRO para realizar todo el trabajo administrativo de un ensayo clínico. Para las fases II a IV, la CRO recluta a los investigadores participantes, los capacita, les proporciona suministros, coordina la administración del estudio y la recopilación de datos , organiza reuniones, monitorea los sitios para cumplir con el protocolo clínico y se asegura de que el patrocinador reciba datos de cada sitio. También se pueden contratar organizaciones especializadas en la gestión de sitios para coordinar con la CRO para garantizar una aprobación rápida del IRB/IEC y una iniciación más rápida del sitio y el reclutamiento de pacientes. Los ensayos clínicos de fase I de nuevos medicamentos a menudo se llevan a cabo en una clínica de ensayos clínicos especializada, con farmacólogos dedicados, donde los sujetos pueden ser observados por personal de tiempo completo. Estas clínicas suelen estar dirigidas por una CRO que se especializa en estos estudios.
En un centro participante, uno o más asistentes de investigación (a menudo, enfermeras) realizan la mayor parte del trabajo de realización del ensayo clínico. El trabajo del asistente de investigación puede incluir algunas o todas las siguientes tareas: proporcionar a la junta de revisión institucional (IRB) local la documentación necesaria para obtener su permiso para realizar el estudio, ayudar con el inicio del estudio, identificar a los pacientes elegibles, obtener el consentimiento de ellos o de sus familias, administrar el tratamiento o tratamientos del estudio, recopilar y analizar estadísticamente los datos, mantener y actualizar los archivos de datos durante el seguimiento y comunicarse con la IRB, así como con el patrocinador y la CRO.
En el contexto de un ensayo clínico, la calidad generalmente se refiere a la ausencia de errores que puedan afectar la toma de decisiones, tanto durante la realización del ensayo como en el uso de sus resultados. [62]
Un modelo de justicia interaccional puede utilizarse para probar los efectos de la voluntad de hablar con un médico sobre la inscripción en un ensayo clínico. [63] Los resultados encontraron que los candidatos potenciales a ensayos clínicos tenían menos probabilidades de inscribirse en ensayos clínicos si el paciente estaba más dispuesto a hablar con su médico. El razonamiento detrás de este descubrimiento puede ser que los pacientes están contentos con su atención actual. Otra razón para la relación negativa entre la justicia percibida y la inscripción en ensayos clínicos es la falta de independencia del proveedor de atención. Los resultados encontraron que existe una relación positiva entre la falta de voluntad de hablar con su médico y la inscripción en ensayos clínicos. La falta de voluntad de hablar sobre ensayos clínicos con los proveedores de atención actuales puede deberse a la independencia de los pacientes con respecto al médico. Los pacientes que tienen menos probabilidades de hablar sobre ensayos clínicos están más dispuestos a utilizar otras fuentes de información para obtener una mejor comprensión de los tratamientos alternativos. La inscripción en ensayos clínicos debe estar motivada para utilizar sitios web y publicidad televisiva para informar al público sobre la inscripción en ensayos clínicos.
En la última década se ha observado una proliferación del uso de la tecnología de la información en la planificación y realización de ensayos clínicos. Los patrocinadores de investigación o las CRO suelen utilizar sistemas de gestión de ensayos clínicos para ayudar a planificar y gestionar los aspectos operativos de un ensayo clínico, en particular con respecto a los centros de investigación. Los análisis avanzados para identificar investigadores y centros de investigación con experiencia en un área determinada utilizan información pública y privada sobre la investigación en curso. [64] La captura electrónica de datos (EDC) basada en la web y los sistemas de gestión de datos clínicos se utilizan en la mayoría de los ensayos clínicos [65] para recopilar datos de informes de casos de los centros, gestionar su calidad y prepararlos para el análisis. Los centros utilizan sistemas de respuesta de voz interactiva para registrar la inscripción de pacientes mediante un teléfono y para asignarlos a un grupo de tratamiento en particular (aunque los teléfonos están siendo reemplazados cada vez más por herramientas basadas en la web (IWRS) que a veces forman parte del sistema EDC). Si bien en el pasado los resultados informados por los pacientes solían basarse en papel, cada vez más las mediciones se recopilan mediante portales web o dispositivos ePRO (o eDiary) portátiles , a veces inalámbricos. [66] Se utiliza software estadístico para analizar los datos recopilados y prepararlos para su presentación reglamentaria. El acceso a muchas de estas aplicaciones se agrega cada vez más en portales de ensayos clínicos basados en la web . En 2011, la FDA aprobó un ensayo de fase I que utilizó telemonitorización, también conocida como monitoreo remoto de pacientes, para recopilar datos biométricos en los hogares de los pacientes y transmitirlos electrónicamente a la base de datos del ensayo. Esta tecnología proporciona muchos más puntos de datos y es mucho más conveniente para los pacientes, porque tienen menos visitas a los sitios de prueba. Como se señala a continuación, los ensayos clínicos descentralizados son aquellos que no requieren la presencia física de los pacientes en un sitio y, en cambio, se basan en gran medida en la recopilación de datos de salud digitales, procesos de consentimiento informado digitales, etc.
Un ensayo clínico produce datos que podrían revelar diferencias cuantitativas entre dos o más intervenciones; se utilizan análisis estadísticos para determinar si dichas diferencias son verdaderas, son resultado del azar o son las mismas que no se obtienen con ningún tratamiento (placebo). [67] [68] Los datos de un ensayo clínico se acumulan gradualmente a lo largo de la duración del ensayo, que puede ir desde meses hasta años. [53] En consecuencia, los resultados de los participantes reclutados al principio del estudio están disponibles para el análisis mientras los sujetos todavía están siendo asignados a los grupos de tratamiento en el ensayo. El análisis temprano puede permitir que la evidencia emergente ayude a tomar decisiones sobre si detener el estudio o reasignar a los participantes al segmento más exitoso del ensayo. [67] Los investigadores también pueden querer detener un ensayo cuando el análisis de datos no muestra ningún efecto del tratamiento. [68]
Los ensayos clínicos son supervisados de cerca por las autoridades regulatorias correspondientes. Todos los estudios que implican una intervención médica o terapéutica en pacientes deben ser aprobados por un comité de ética supervisor antes de que se otorgue el permiso para realizar el ensayo. El comité de ética local tiene discreción sobre cómo supervisará los estudios no intervencionistas (estudios observacionales o aquellos que utilizan datos ya recopilados). En los EE. UU., este organismo se denomina Junta de Revisión Institucional (IRB); en la UE, se denominan comités de ética . La mayoría de los IRB están ubicados en el hospital o institución del investigador local, pero algunos patrocinadores permiten el uso de un IRB central (independiente/con fines de lucro) para los investigadores que trabajan en instituciones más pequeñas.
Para ser éticos, los investigadores deben obtener el consentimiento pleno e informado de los sujetos humanos participantes. (Una de las principales funciones del IRB es garantizar que los pacientes potenciales estén adecuadamente informados sobre el ensayo clínico). Si el paciente no puede dar su consentimiento por sí mismo, los investigadores pueden solicitar el consentimiento del representante legalmente autorizado del paciente. Además, los participantes del ensayo clínico deben saber que pueden retirarse del ensayo clínico en cualquier momento sin que se tomen medidas adversas en su contra. [69] En California , el estado ha priorizado a las personas que pueden actuar como representante legalmente autorizado. [70]
En algunos lugares de los EE. UU., la IRB local debe certificar a los investigadores y a su personal antes de que puedan realizar ensayos clínicos. Deben comprender la ley federal de privacidad del paciente ( HIPAA ) y las buenas prácticas clínicas. La Conferencia Internacional de Armonización de las Directrices para las Buenas Prácticas Clínicas es un conjunto de estándares utilizados internacionalmente para la realización de ensayos clínicos. Las directrices tienen como objetivo garantizar que "se protejan los derechos, la seguridad y el bienestar de los sujetos de los ensayos".
El concepto de consentimiento informado de los sujetos humanos participantes existe en muchos países, pero su definición precisa aún puede variar.
El consentimiento informado es claramente una condición "necesaria" para la conducta ética, pero no "garantiza" la conducta ética. En los ensayos de uso compasivo , esto último se convierte en un problema particularmente difícil. El objetivo final es servir a la comunidad de pacientes o futuros pacientes de la mejor manera posible y de la manera más responsable. Véase también Acceso ampliado . Sin embargo, puede resultar difícil convertir este objetivo en una función objetiva cuantificada y bien definida. Sin embargo, en algunos casos esto se puede hacer, por ejemplo, para las preguntas sobre cuándo detener los tratamientos secuenciales (véase Algoritmo de probabilidades ), y entonces los métodos cuantificados pueden desempeñar un papel importante.
Existen preocupaciones éticas adicionales cuando se realizan ensayos clínicos en niños ( pediatría ) y en situaciones de emergencia o epidemia. [71] [72]
Puede resultar difícil equilibrar éticamente los derechos de múltiples partes interesadas. Por ejemplo, cuando fracasan los ensayos de medicamentos, los patrocinadores pueden tener el deber de informar de inmediato a los inversores actuales y potenciales, lo que significa que tanto el personal de investigación como los participantes inscritos pueden enterarse por primera vez del final de un ensayo a través de noticias comerciales públicas . [73]
En respuesta a casos específicos en los que no se publicaron datos desfavorables de investigaciones patrocinadas por compañías farmacéuticas, la Asociación de Fabricantes e Investigadores Farmacéuticos de Estados Unidos publicó nuevas directrices que instan a las compañías a informar todos los hallazgos y limitar la participación financiera de los investigadores en las compañías farmacéuticas. [74] El Congreso de los EE. UU. firmó una ley que requiere que los ensayos clínicos de fase II y fase III sean registrados por el patrocinador en el sitio web clinicaltrials.gov compilado por los Institutos Nacionales de Salud . [75]
Los investigadores de fármacos que no son empleados directamente por las compañías farmacéuticas a menudo buscan subvenciones de los fabricantes, y los fabricantes a menudo recurren a investigadores académicos para realizar estudios dentro de las redes de universidades y sus hospitales, por ejemplo, para la investigación traslacional del cáncer. De manera similar, la competencia por puestos académicos titulares, subvenciones gubernamentales y prestigio crea conflictos de intereses entre los científicos académicos. [76] Según un estudio, aproximadamente el 75% de los artículos retractados por razones relacionadas con la mala conducta no cuentan con apoyo financiero declarado de la industria. [77] Los ensayos de siembra son particularmente controvertidos. [78]
En los Estados Unidos, todos los ensayos clínicos presentados ante la FDA como parte de un proceso de aprobación de medicamentos son evaluados independientemente por expertos clínicos dentro de la Administración de Alimentos y Medicamentos, [79] incluidas las inspecciones de la recopilación de datos primarios en sitios de ensayos clínicos seleccionados. [80]
En 2001, los editores de 12 importantes revistas publicaron un editorial conjunto, publicado en cada revista, sobre el control que ejercen los patrocinadores sobre los ensayos clínicos, en particular el uso de contratos que permiten a los patrocinadores revisar los estudios antes de su publicación y no publicarlos. Para contrarrestar este efecto, reforzaron las restricciones editoriales. El editorial señalaba que, en 2000, las organizaciones de investigación por contrato habían recibido el 60% de las subvenciones de las empresas farmacéuticas en los Estados Unidos. A los investigadores se les puede restringir la contribución al diseño de los ensayos, el acceso a los datos brutos y la interpretación de los resultados. [81]
A pesar de las recomendaciones explícitas de las partes interesadas sobre medidas para mejorar los estándares de la investigación médica patrocinada por la industria, [82] en 2013, Tohen advirtió sobre la persistencia de una brecha en la credibilidad de las conclusiones derivadas de los ensayos clínicos financiados por la industria, y pidió garantizar una estricta adhesión a las normas éticas en las colaboraciones industriales con la academia, a fin de evitar una mayor erosión de la confianza del público. [83] Las cuestiones a las que se hace referencia para su atención a este respecto incluyen el posible sesgo de observación, la duración del tiempo de observación para los estudios de mantenimiento, la selección de las poblaciones de pacientes, los factores que afectan la respuesta al placebo y las fuentes de financiación. [84] [85] [86]
La realización de ensayos clínicos de vacunas durante epidemias y pandemias está sujeta a preocupaciones éticas. En el caso de enfermedades con altas tasas de mortalidad, como el ébola, asignar a los individuos a un grupo placebo o de control puede considerarse una sentencia de muerte. En respuesta a las preocupaciones éticas sobre la investigación clínica durante epidemias, la Academia Nacional de Medicina redactó un informe en el que se identifican siete consideraciones éticas y científicas, a saber: [87]
Las mujeres embarazadas y los niños suelen quedar excluidos de los ensayos clínicos por ser poblaciones vulnerables, aunque los datos que apoyan esta exclusión no son sólidos. Al excluirlos de los ensayos clínicos, a menudo se carece de información sobre la seguridad y la eficacia de las terapias para estas poblaciones. Durante los inicios de la epidemia del VIH/SIDA , un científico observó que al excluir a estos grupos de un tratamiento que podría salvarles la vida, se los estaba "protegiendo hasta la muerte". Proyectos como el de Ética de la investigación para vacunas, epidemias y nuevas tecnologías (PREVENT) han abogado por la inclusión ética de las mujeres embarazadas en los ensayos de vacunas. La inclusión de los niños en los ensayos clínicos tiene consideraciones morales adicionales, ya que los niños carecen de autonomía para tomar decisiones. En el pasado, se había criticado a los ensayos por utilizar niños hospitalizados o huérfanos; estas preocupaciones éticas detuvieron de hecho las investigaciones futuras. En un esfuerzo por mantener una atención pediátrica eficaz, varios países europeos y los EE. UU. tienen políticas para incitar u obligar a las empresas farmacéuticas a realizar ensayos pediátricos. Las directrices internacionales recomiendan ensayos pediátricos éticos limitando el daño, considerando los diversos riesgos y teniendo en cuenta las complejidades de la atención pediátrica. [87]
La responsabilidad por la seguridad de los sujetos en un ensayo clínico se comparte entre el patrocinador, los investigadores del sitio local (si son diferentes del patrocinador), los diversos IRB que supervisan el estudio y (en algunos casos, si el estudio involucra un medicamento o dispositivo comercializable), la agencia reguladora del país donde se venderá el medicamento o dispositivo.
Con frecuencia se emplea una revisión de seguridad concurrente sistemática para garantizar la seguridad de los participantes en la investigación. La realización y la revisión continua están diseñadas para ser proporcionales al riesgo del ensayo. Por lo general, esta función la desempeña un Comité de Datos y Seguridad , un Monitor de Seguridad Médica designado externamente, [88] un Oficial de Seguridad Independiente o, en el caso de estudios pequeños o de bajo riesgo, el investigador principal. [89]
Por razones de seguridad, muchos ensayos clínicos de medicamentos [90] están diseñados para excluir a las mujeres en edad fértil, las mujeres embarazadas o las mujeres que quedan embarazadas durante el estudio. En algunos casos, también se excluye a las parejas masculinas de estas mujeres o se les exige que tomen medidas de control de la natalidad.
Durante todo el ensayo clínico, el patrocinador es responsable de informar con precisión a los investigadores del centro local sobre el verdadero historial de seguridad del fármaco, dispositivo u otros tratamientos médicos que se van a probar, y sobre cualquier interacción potencial de los tratamientos del estudio con tratamientos ya aprobados. Esto permite a los investigadores locales tomar una decisión informada sobre si participar en el estudio o no. El patrocinador también es responsable de monitorear los resultados del estudio a medida que llegan de los diversos sitios a medida que avanza el ensayo. En ensayos clínicos más grandes, un patrocinador utilizará los servicios de un comité de monitoreo de datos (DMC, conocido en los EE. UU. como una junta de monitoreo de seguridad de datos). Este grupo independiente de médicos y estadísticos se reúne periódicamente para revisar los datos no ciegos que el patrocinador ha recibido hasta el momento. El DMC tiene el poder de recomendar la terminación del estudio en función de su revisión, por ejemplo, si el tratamiento del estudio está causando más muertes que el tratamiento estándar o parece estar causando eventos adversos graves inesperados y relacionados con el estudio . El patrocinador es responsable de recopilar informes de eventos adversos de todos los investigadores del sitio del estudio y de informar a todos los investigadores sobre el criterio del patrocinador sobre si estos eventos adversos estaban relacionados o no con el tratamiento del estudio.
El patrocinador y los investigadores del centro local son responsables conjuntamente de redactar un consentimiento informado específico del centro que informe con precisión a los posibles sujetos sobre los verdaderos riesgos y los posibles beneficios de participar en el estudio, al mismo tiempo que presenta el material de la forma más breve posible y en un lenguaje corriente. Las normas de la FDA establecen que la participación en ensayos clínicos es voluntaria y que el sujeto tiene derecho a no participar o a dejar de participar en cualquier momento. [91]
El principio ético de primum non-nocere ("lo primero es no hacer daño") guía el ensayo y, si un investigador cree que el tratamiento del estudio puede estar dañando a los sujetos del estudio, puede dejar de participar en cualquier momento. Por otro lado, los investigadores a menudo tienen un interés financiero en reclutar sujetos y podrían actuar de manera poco ética para obtener y mantener su participación.
Los investigadores locales son responsables de llevar a cabo el estudio de acuerdo con el protocolo del estudio y de supervisar al personal del estudio durante toda la duración del mismo. El investigador local o su personal del estudio también son responsables de garantizar que los posibles sujetos del estudio comprendan los riesgos y los posibles beneficios de participar en el estudio. En otras palabras, ellos (o sus representantes legalmente autorizados) deben dar un consentimiento verdaderamente informado.
Los investigadores locales son responsables de revisar todos los informes de eventos adversos enviados por el patrocinador. Estos informes de eventos adversos contienen las opiniones tanto del investigador (en el sitio donde ocurrió el evento adverso) como del patrocinador, con respecto a la relación del evento adverso con los tratamientos del estudio. Los investigadores locales también son responsables de emitir un juicio independiente sobre estos informes e informar de inmediato al IRB local sobre todos los eventos adversos graves y relacionados con el tratamiento del estudio.
Cuando un investigador local es el patrocinador, es posible que no haya informes formales de eventos adversos, pero el personal del estudio en todas las ubicaciones es responsable de informar al investigador coordinador sobre cualquier cosa inesperada. El investigador local es responsable de ser sincero con el comité de revisión institucional local en todas las comunicaciones relacionadas con el estudio.
La aprobación de una Junta de Revisión Institucional (IRB, por sus siglas en inglés) o un Comité de Ética Independiente (IEC, por sus siglas en inglés) es necesaria antes de que se pueda comenzar cualquier investigación, excepto la más informal. En los ensayos clínicos comerciales, el protocolo del estudio no es aprobado por una IRB antes de que el patrocinador reclute a los centros para realizar el ensayo. Sin embargo, el protocolo y los procedimientos del estudio se han adaptado para cumplir con los requisitos genéricos de presentación de informes de la IRB. En este caso, y cuando no hay un patrocinador independiente, cada investigador del centro local presenta el protocolo del estudio, los consentimientos, los formularios de recopilación de datos y la documentación de respaldo a la IRB local. Las universidades y la mayoría de los hospitales tienen IRB internos. Otros investigadores (como en las clínicas sin cita previa) utilizan IRB independientes.
El IRB examina el estudio tanto para garantizar su seguridad médica como para proteger a los pacientes que participan en él antes de permitir que el investigador comience el estudio. Puede exigir cambios en los procedimientos del estudio o en las explicaciones que se dan al paciente. Un informe anual obligatorio de "revisión continua" del investigador actualiza al IRB sobre el progreso del estudio y cualquier nueva información de seguridad relacionada con el estudio.
En los EE.UU., la FDA puede auditar los archivos de los investigadores de los centros locales después de que hayan terminado de participar en un estudio, para ver si estaban siguiendo correctamente los procedimientos del estudio. Esta auditoría puede ser aleatoria o por una causa (porque se sospecha que el investigador ha proporcionado datos fraudulentos). Evitar una auditoría es un incentivo para que los investigadores sigan los procedimientos del estudio. Un "estudio clínico cubierto" se refiere a un ensayo presentado a la FDA como parte de una solicitud de comercialización (por ejemplo, como parte de una NDA o 510(k) ), sobre el cual la FDA puede exigir la divulgación del interés financiero del investigador clínico en el resultado del estudio. Por ejemplo, el solicitante debe revelar si un investigador posee acciones del patrocinador o si posee un interés de propiedad en el producto bajo investigación. La FDA define un estudio cubierto como "... cualquier estudio de un fármaco, producto biológico o dispositivo en humanos presentado en una solicitud de comercialización o petición de reclasificación en el que el solicitante o la FDA se basan para establecer que el producto es eficaz (incluidos los estudios que muestran equivalencia con un producto eficaz) o cualquier estudio en el que un solo investigador hace una contribución significativa a la demostración de seguridad". [92]
Por otra parte, muchas compañías farmacéuticas estadounidenses han trasladado algunos ensayos clínicos al extranjero. Entre las ventajas de realizar ensayos en el extranjero se encuentran los menores costes (en algunos países) y la posibilidad de realizar ensayos más amplios en plazos más cortos, mientras que existe una posible desventaja en la gestión de ensayos de menor calidad. [93] Los distintos países tienen diferentes requisitos regulatorios y capacidades de aplicación. Se estima que actualmente el 40% de todos los ensayos clínicos se llevan a cabo en Asia, Europa del Este y América Central y del Sur. "No existe un sistema de registro obligatorio para los ensayos clínicos en estos países y muchos no siguen las directivas europeas en sus operaciones", dice Jacob Sijtsma de WEMOS, una organización de defensa de la salud con sede en los Países Bajos que hace un seguimiento de los ensayos clínicos en los países en desarrollo. [94]
A partir de la década de 1980, se demostró que la armonización de los protocolos de ensayos clínicos era factible en todos los países de la Unión Europea. Al mismo tiempo, la coordinación entre Europa, Japón y los Estados Unidos condujo a una iniciativa conjunta de la industria y los organismos reguladores sobre la armonización internacional, que en 1990 recibió el nombre de Conferencia Internacional sobre Armonización de los Requisitos Técnicos para el Registro de Productos Farmacéuticos para Uso Humano (ICH) [95]. En la actualidad, la mayoría de los programas de ensayos clínicos siguen las directrices de la ICH, cuyo objetivo es "garantizar que se desarrollen y registren medicamentos de buena calidad, seguros y eficaces de la manera más eficiente y rentable. Estas actividades se llevan a cabo en beneficio del consumidor y de la salud pública, para evitar la duplicación innecesaria de ensayos clínicos en seres humanos y para minimizar el uso de pruebas con animales sin comprometer las obligaciones regulatorias de seguridad y eficacia". [96]
La agregación de datos de seguridad de los ensayos clínicos durante el desarrollo de un fármaco es importante porque los ensayos suelen estar diseñados para centrarse en determinar qué tan bien funciona el fármaco. Los datos de seguridad recopilados y agregados en múltiples ensayos a medida que se desarrolla el fármaco permiten al patrocinador, a los investigadores y a las agencias reguladoras monitorear el perfil de seguridad agregado de los medicamentos experimentales a medida que se desarrollan. El valor de evaluar los datos de seguridad agregados es que: a) las decisiones basadas en la evaluación de seguridad agregada durante el desarrollo del fármaco se pueden tomar durante todo el desarrollo del fármaco y b) prepara al patrocinador y a los reguladores para evaluar la seguridad del fármaco después de que se apruebe. [97] [98] [99] [100] [101]
Los costos de los ensayos clínicos varían según la fase del ensayo, el tipo de ensayo y la enfermedad estudiada. Un estudio de ensayos clínicos realizado en los Estados Unidos entre 2004 y 2012 determinó que el costo promedio de los ensayos de Fase I oscilaba entre $1,4 millones y $6,6 millones, según el tipo de enfermedad. Los ensayos de Fase II oscilaban entre $7 millones y $20 millones, y los de Fase III entre $11 millones y $53 millones. [102]
El costo de un estudio depende de muchos factores, especialmente la cantidad de sitios que lo realizan, la cantidad de pacientes involucrados y si el tratamiento del estudio ya está aprobado para uso médico.
Los gastos en que incurra una compañía farmacéutica al administrar un ensayo clínico de fase III o IV pueden incluir, entre otros:
Estos gastos se generan a lo largo de varios años.
En los Estados Unidos, los patrocinadores pueden recibir un crédito fiscal del 50 por ciento por los ensayos clínicos realizados sobre medicamentos que se están desarrollando para el tratamiento de enfermedades huérfanas . [103] Las agencias nacionales de salud, como los Institutos Nacionales de Salud de los Estados Unidos , ofrecen subvenciones a los investigadores que diseñan ensayos clínicos que intentan responder a preguntas de investigación de interés para la agencia. En estos casos, el investigador que escribe la subvención y administra el estudio actúa como patrocinador y coordina la recopilación de datos de otros sitios. Estos otros sitios pueden o no recibir un pago por participar en el estudio, dependiendo del monto de la subvención y la cantidad de esfuerzo que se espera de ellos. El uso de recursos de Internet puede, en algunos casos, reducir la carga económica. [104]
Los investigadores suelen recibir una compensación por su trabajo en los ensayos clínicos. Estas cantidades pueden ser pequeñas, y cubrir solo un salario parcial de los asistentes de investigación y el costo de los suministros (normalmente es el caso de los estudios de las agencias nacionales de salud), o pueden ser sustanciales e incluir los "gastos generales" que permiten al investigador pagar al personal de investigación durante los períodos entre los ensayos clínicos. [ cita requerida ]
Los participantes en los ensayos de medicamentos de fase I no obtienen ningún beneficio directo para la salud por participar. Por lo general, se les paga un honorario por su tiempo, con pagos regulados y no relacionados con ningún riesgo involucrado. Las motivaciones de los voluntarios sanos no se limitan a la recompensa financiera y pueden incluir otras motivaciones como contribuir a la ciencia y otras. [105] En los ensayos de fase posterior, es posible que no se pague a los sujetos para asegurar su motivación para participar con potencial para un beneficio de salud o contribuir al conocimiento médico. Se pueden realizar pequeños pagos para gastos relacionados con el estudio, como viajes o como compensación por su tiempo para proporcionar información de seguimiento sobre su salud después de que finalice el tratamiento del ensayo.

Los ensayos de fármacos de fase 0 y fase I buscan voluntarios sanos. La mayoría de los demás ensayos clínicos buscan pacientes que tengan una enfermedad o afección médica específica. La diversidad observada en la sociedad debería reflejarse en los ensayos clínicos mediante la inclusión adecuada de poblaciones de minorías étnicas . [106] El reclutamiento de pacientes o de participantes desempeña un papel importante en las actividades y responsabilidades de los centros que realizan ensayos clínicos. [107]
Todos los voluntarios que se consideren para un ensayo deben someterse a un examen médico. Los requisitos varían según las necesidades del ensayo, pero por lo general los voluntarios se someterán a exámenes en un laboratorio médico para: [108]
Se ha observado que los participantes en ensayos clínicos son desproporcionadamente blancos. [109] [110] A menudo, las minorías no están informadas sobre los ensayos clínicos. [111] Una revisión sistemática reciente de la literatura encontró que la raza/etnia, así como el sexo, no estaban bien representados ni, a veces, ni siquiera se los rastreaba como participantes en una gran cantidad de ensayos clínicos sobre el manejo de la pérdida auditiva en adultos. [112] Esto puede reducir la validez de los hallazgos con respecto a los pacientes no blancos [113] al no representar adecuadamente a la población más grande .
Según el tipo de participantes requeridos, los patrocinadores de los ensayos clínicos, o las organizaciones de investigación por contrato que trabajan en su nombre, tratan de encontrar sitios con personal calificado, así como acceso a pacientes que podrían participar en el ensayo. Al trabajar con esos sitios, pueden utilizar diversas estrategias de reclutamiento, incluidas bases de datos de pacientes, anuncios en periódicos y radio, folletos, carteles en lugares a los que los pacientes podrían ir (como consultorios médicos) y reclutamiento personal de pacientes por parte de los investigadores.
Los voluntarios con enfermedades o afecciones específicas tienen recursos adicionales en línea que los ayudan a localizar ensayos clínicos. Por ejemplo, el Fox Trial Finder conecta los ensayos de la enfermedad de Parkinson en todo el mundo con voluntarios que tienen un conjunto específico de criterios, como ubicación, edad y síntomas. [114] Existen otros servicios específicos de la enfermedad para que los voluntarios encuentren ensayos relacionados con su afección. [115] Los voluntarios pueden buscar directamente en ClinicalTrials.gov para localizar ensayos utilizando un registro administrado por los Institutos Nacionales de Salud y la Biblioteca Nacional de Medicina de los EE. UU . También existe un software que permite a los médicos encontrar opciones de ensayos para un paciente individual en función de datos como los datos genómicos. [116]

El modelo de búsqueda y procesamiento de información sobre riesgos (RISP) analiza las implicaciones sociales que afectan las actitudes y la toma de decisiones en relación con los ensayos clínicos. [117] Las personas que tienen un mayor interés en el tratamiento proporcionado en un ensayo clínico mostraron una mayor probabilidad de buscar información sobre los ensayos clínicos. Los pacientes con cáncer manifestaron actitudes más optimistas hacia los ensayos clínicos que la población general. Tener una perspectiva más optimista sobre los ensayos clínicos también conduce a una mayor probabilidad de participar. [117]
El emparejamiento implica una comparación sistemática de la información clínica y demográfica de un paciente con respecto a los criterios de elegibilidad de varios ensayos. Los métodos incluyen:
Aunque los ensayos se realizan habitualmente en los principales centros médicos, algunos participantes quedan excluidos debido a la distancia y los gastos necesarios para el viaje, lo que genera dificultades, desventajas y desigualdad para los participantes, especialmente aquellos de comunidades rurales y desatendidas. Por lo tanto, el concepto de un "ensayo clínico descentralizado" que minimiza o elimina la necesidad de que los pacientes viajen a los sitios, [122] ahora está más extendido, una capacidad mejorada por la telesalud y las tecnologías portátiles . [123]
De los medicamentos que se han iniciado en ensayos clínicos en humanos, solo el 10 por ciento obtiene la aprobación de la FDA.
...
{{cite journal}}: CS1 maint: DOI inactivo a partir de marzo de 2024 ( enlace )