Teoría del estado de transición
[6] Por lo tanto, fue necesario un desarrollo posterior para comprender los dos parámetros asociados con esta ley, el factor preexponencial (A) y la energía de activación (Ea).
es llamada factor de frecuencia o coeficiente preexponencial, y
Aunque a comienzos del siglo XX la ecuación de Arrhenius era comúnmente aceptada, la interpretación física de A y E resultaba vaga.
Esto llevó a muchos investigadores en cinética química a ofrecer diferentes teorías sobre cómo ocurrían las reacciones químicas, en un intento de relacionar A y E con la dinámica molecular directamente responsable de las reacciones químicas.
A la vez que Marcelin desarrollaba su formulación, los químicos holandeses Philip Abraham Kohnstamm, Frans Eppo Cornelis Scheffer, y Wiedold Frans Brandsma introdujeron por vez primera la entropía estándar de activación y la entalpía estándar de activación.
La naturaleza o significado de la constante seguía sin estar clara.
Lewis aplicó su tratamiento a la siguiente reacción y obtuvo una buena concordancia con los resultados experimentales.
Sin embargo, más tarde aplicó el mismo tratamiento a otras reacciones y encontró discrepancias grandes entre resultados teóricos y experimentales .
Sin embargo, la aplicación de la Mecánica estadística a la teoría del estado de transición se desarrolló muy lentamente porque, a mediados del siglo XIX, James Clerk Maxwell, Ludwig Boltzmann, y Leopold Pfaundler publicaron varios artículos discutiendo las reacciones en equilibrio y las velocidades de reacción, en términos del movimiento molecular y la distribución estadística de las velocidades moleculares.
donde a y b son constantes relacionadas con términos de energía.
Luego aplicó los procedimientos mecánico-estadísticos de Gibbs y obtuvo una expresión similar a la que había obtenido anteriormente a partir de consideraciones termodinámicas.
En 1915, otra importante contribución fue realizada por el físico británico James Rice.
Basados en su análisis estadístico, concluyó que la constante de velocidad es proporcional al "incremento crítico".
Obtuvo la siguiente ecuación para la constante de velocidad de la reacción directa: donde: Esta expresión es muy importante ya que es la primera vez que el factor
En 1920, el químico americano Richard Chase Tolman desarrolló adicionalmente la idea de Rice del "incremento crítico".
En 1931, Eyring y Polanyi construyeron una superficie de energía potencial para la reacción inferior.
Esta superficie es un diagrama tridimensional basado en los principios mecano-cuánticos así como en datos experimentales de frecuencias vibracionales y energías de disociación.
La única característica importante añadida por Eyring, Polanyi y Evans fue la noción de que los complejos activados están en cuasi-equilibrio con los reactivos.
La velocidad es por tanto directamente proporcional a la concentración de esos complejos multiplicado por la frecuencia (kBT/h) con la que se convierten en productos.
donde se ha alcanzado el equilibrio entre todas las especies del sistema incluyendo los complejos activados, [AB]‡ .
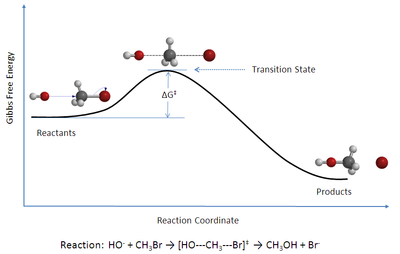
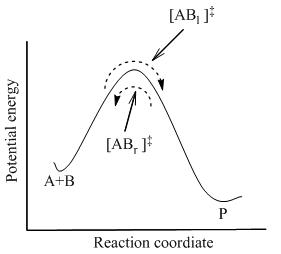
La teoría del estado de transición postula que aun cuando los reactivos y productos no estén en equilibrio entre sí, los complejos activados están en cuasi-equilibrio con los reactivos.
Si las moléculas de producto son frecuentemente retiradas del sistema reaccionante, el flujo de esos complejos activados que comenzaron como productos ([ABr]‡ ) se detendrá; sin embargo, todavía habrá un flujo de izquierda a derecha.
En la teoría del estado de transición, es importante darse cuenta de que cuando se dice que los complejos activados están en equilibrio con los reactivos, se refiere solamente a aquellos complejos activados ([ABl] ‡) que eran moléculas de reactivo en un estado inmediato.
k‡ es directamente proporcional a la frecuencia del modo vibracional responsable de convertir el complejo activado en producto; la frecuencia de este modo vibracional es ν.
En general, la teoría del estado de transición ha suministrado a los investigadores una base conceptual para comprender cómo tienen lugar las reacciones químicas.
[8] Se supone que a no ser que los átomos o las moléculas colisionen con suficiente energía para formar la estructura de transición, o bien la reacción no ocurrirá.
Con respecto a las reacciones químicas esto significa que hay una oportunidad de que las moléculas reaccionen incluso si no colisionan con suficiente energía para atravesar la barrera de energía.
La teoría del estado de transición falla para algunas reacciones a alta temperatura.
Mientras esta descripción es consistente para reacciones que ocurren a temperaturas relativamente bajas, a altas temperaturas, las moléculas se distribuyen en los modos de mayor energía vibracional; su movimiento se hace más complejo y las colisiones pueden conducir a estados de transición alejados de lo previsto por la teoría del estado de transición.
[10] Dadas estas limitaciones, se han propuesto varias alternativas a la teoría del estado de transición, como las que siguen.