Las talasemias son trastornos sanguíneos hereditarios que dan lugar a una hemoglobina anormal . [7] Los síntomas dependen del tipo de talasemia y pueden variar de ninguno a grave. [1] A menudo hay anemia de leve a grave (nivel bajo de glóbulos rojos o hemoglobina), ya que la talasemia puede afectar la producción de glóbulos rojos y también afectar la duración de la vida de los glóbulos rojos. [1] Los síntomas de anemia incluyen sensación de cansancio y palidez de la piel . [1] Otros síntomas de la talasemia incluyen problemas óseos, agrandamiento del bazo , piel amarillenta , hipertensión pulmonar y orina oscura. [1] Puede producirse un crecimiento lento en los niños. [1] Los síntomas y presentaciones de la talasemia pueden cambiar con el tiempo. Los términos más antiguos incluían anemia de Cooley y anemia mediterránea para la beta-talasemia. Estos han sido reemplazados por los términos talasemia dependiente de transfusión (TDT) y talasemia no dependiente de transfusión (NTDT). Los pacientes con TDT requieren transfusiones regulares, normalmente cada dos a cinco semanas. Las TDT incluyen beta-talasemia mayor, enfermedad de HbH no delecional, enfermedad de Bart con Hb sobreviviente y HbE/beta-talasemia grave. [8]
Las talasemias son trastornos genéticos . [2] Hay dos tipos principales, talasemia alfa y talasemia beta . [7] La gravedad de la talasemia alfa y beta depende de cuántos de los cuatro genes de la globina alfa o dos genes de la globina beta faltan. [2] El diagnóstico se realiza típicamente mediante análisis de sangre que incluyen un hemograma completo , pruebas especiales de hemoglobina y pruebas genéticas. [3] El diagnóstico puede ocurrir antes del nacimiento a través de pruebas prenatales . [9]
El tratamiento depende del tipo y la gravedad. [4] El tratamiento para aquellos con enfermedad más grave a menudo incluye transfusiones de sangre regulares , quelación de hierro y ácido fólico . [4] La quelación de hierro se puede realizar con deferoxamina , deferasirox o deferiprona . [4] [10] Ocasionalmente, un trasplante de médula ósea puede ser una opción. [4] Las complicaciones pueden incluir sobrecarga de hierro de las transfusiones con la consiguiente enfermedad cardíaca o hepática , infecciones y osteoporosis . [1] Si el bazo se agranda demasiado, puede ser necesaria la extirpación quirúrgica . [1] Los pacientes con talasemia que no responden bien a las transfusiones de sangre pueden tomar hidroxiurea o talidomida y, a veces, una combinación de ambas. [11] La hidroxiurea es el único fármaco aprobado por la FDA para la talasemia. Los pacientes que tomaron 10 mg/kg de hidroxiurea todos los días durante un año tuvieron niveles de hemoglobina significativamente más altos, y fue un tratamiento bien tolerado por los pacientes que no respondieron bien a las transfusiones de sangre. [12] Otros inductores de hemoglobina conocidos incluyen la talidomida, pero no se ha probado en un entorno clínico. La combinación de talidomida e hidroxiurea resultó en un aumento significativo de los niveles de hemoglobina en pacientes dependientes y no dependientes de transfusiones [13]
A partir de 2015, la talasemia se presenta en alrededor de 280 millones de personas, con alrededor de 439.000 con enfermedad grave. [14] Es más común entre personas de ascendencia griega , italiana , de Oriente Medio , del sur de Asia y africana . [7] Los hombres y las mujeres tienen tasas similares de enfermedad. [ cita requerida ] Resultó en 16.800 muertes en 2015, por debajo de las 36.000 muertes en 1990. [6] [15] Aquellos que tienen grados menores de talasemia, en común con aquellos que tienen el rasgo de células falciformes , tienen cierta protección contra la malaria , lo que explica por qué el rasgo de células falciformes y la talasemia son más comunes en regiones del mundo donde el riesgo de malaria es mayor. [16] Se estima que 1/3 de las personas con talasemia tienen "talasemia no dependiente de transfusiones" y no dependen de transfusiones de sangre regulares y continuas para sobrevivir.
Signos y síntomas
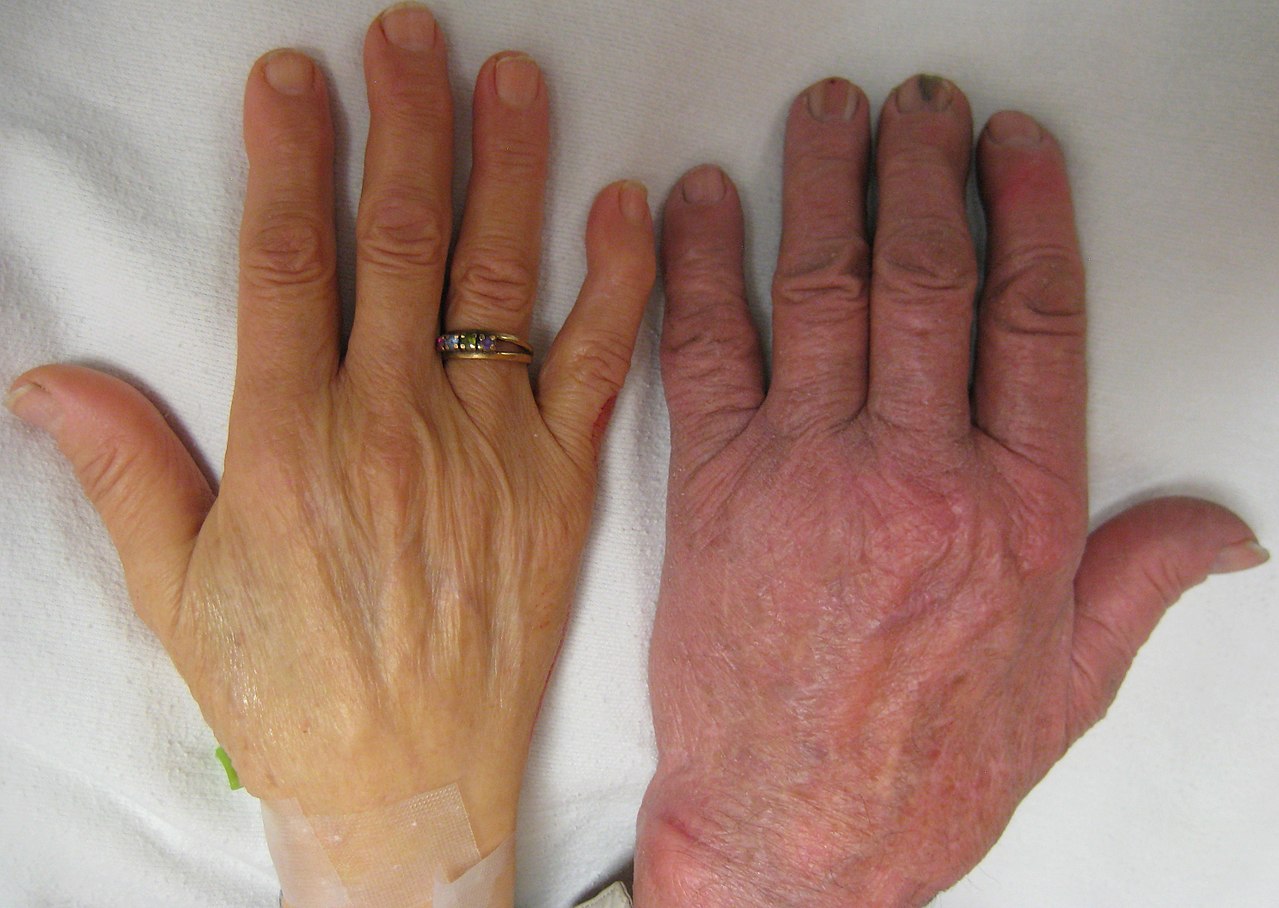
Izquierda: Mano de una persona con anemia severa. Derecha: Mano de una persona sin anemia.Un paciente con talasemia presenta un bazo agrandado.
Sobrecarga de hierro : las personas con talasemia pueden sufrir una sobrecarga de hierro en sus cuerpos, ya sea por la propia enfermedad o por transfusiones de sangre frecuentes. Demasiado hierro puede provocar daños en el corazón, el hígado y el sistema endocrino , que incluye glándulas que producen hormonas que regulan procesos en todo el cuerpo. El daño se caracteriza por depósitos excesivos de hierro. Sin una terapia de quelación de hierro adecuada, casi todos los pacientes con beta-talasemia acumulan niveles de hierro potencialmente fatales. [17]
Infección: Las personas con talasemia tienen un mayor riesgo de contraer infecciones, especialmente si se les ha extirpado el bazo. [18]
Deformidades óseas: la talasemia puede hacer que la médula ósea se expanda, lo que hace que los huesos se ensanchen. Esto puede dar lugar a una estructura ósea anormal, especialmente en la cara y el cráneo. La expansión de la médula ósea también hace que los huesos se vuelvan delgados y quebradizos, lo que aumenta el riesgo de fracturas. [19]
Agrandamiento del bazo : el bazo ayuda a combatir las infecciones y filtra el material no deseado, como los glóbulos rojos viejos o dañados. La talasemia suele ir acompañada de la destrucción de una gran cantidad de glóbulos rojos y la tarea de eliminarlos hace que el bazo se agrande. La esplenomegalia puede empeorar la anemia y puede reducir la vida de los glóbulos rojos transfundidos. Un agrandamiento grave del bazo puede hacer necesaria su extirpación. [20]
Tasas de crecimiento más lentas: la anemia puede hacer que el crecimiento de un niño se ralentice. La pubertad también puede retrasarse en los niños con talasemia. [21]
Problemas cardíacos: Enfermedades como insuficiencia cardíaca congestiva y ritmos cardíacos anormales pueden estar asociadas con talasemia grave. [22]
Biología estructural de la hemoglobina
Las hemoglobinas humanas normales son proteínas tetraméricas compuestas por dos pares de cadenas de globina, cada una de las cuales contiene una cadena de tipo alfa (similar a α) y una cadena de tipo beta (similar a β). Cada cadena de globina está asociada con una fracción de hemo que contiene hierro. A lo largo de la vida, la síntesis de las cadenas de tipo alfa y de tipo beta (también llamadas no similares a alfa) está equilibrada de modo que su proporción es relativamente constante y no hay exceso de ninguno de los dos tipos. [23]
Las cadenas específicas de tipo alfa y beta que se incorporan a la Hb están altamente reguladas durante el desarrollo:
Las Hb embrionarias se expresan ya a las cuatro o seis semanas de embriogénesis y desaparecen alrededor de la octava semana de gestación al ser reemplazadas por Hb fetal. [24] [25] Las Hb embrionarias incluyen:
Hb Gower-1, compuesta por dos globinas ζ (globinas zeta) y dos globinas ε (globinas épsilon) (ζ2ε2)
Hb Gower-2, compuesta por dos globinas alfa y dos globinas épsilon (α2ε2)
Hb Portland I, compuesta por dos globinas zeta y dos globinas gamma (ζ2γ2)
Hb Portland II, compuesta por dos globinas zeta y dos globinas beta (ζ2β2)
La Hb fetal (Hb F) se produce aproximadamente desde la octava semana de gestación hasta el nacimiento y constituye aproximadamente el 80 por ciento de la Hb en el neonato a término. Disminuye durante los primeros meses de vida y, en estado normal, constituye <1 por ciento de la Hb total en la primera infancia y después de ella. La Hb F está compuesta por dos globinas alfa y dos globinas gamma (α2γ2). Los pacientes con β-talasemia presentan niveles más altos de gammaglobulina y, por lo tanto, una mayor producción de Hb F para contrarrestar el desequilibrio que supone no poder producir cadenas beta. [26]
La Hb adulta ( Hb A ) se produce en niveles bajos durante la vida embrionaria y fetal y es la Hb predominante en niños a partir de los seis meses de edad; constituye el 96-97% de la Hb total en individuos sin hemoglobinopatía. Está compuesta por dos globinas alfa y dos globinas beta (α2β2). [ cita requerida ]
La Hb A2 es una Hb menor en adultos que normalmente representa aproximadamente el 2,5-3,5 % de la Hb total a partir de los seis meses de edad. Está compuesta por dos globinas alfa y dos globinas delta (α2δ2). [ cita requerida ]
Tanto las talasemias α como las β suelen heredarse de forma autosómica recesiva . Se han descrito casos de talasemias α y β de herencia dominante , el primero de los cuales se produjo en una familia irlandesa con dos deleciones de 4 y 11 pb en el exón 3 interrumpidas por una inserción de 5 pb en el gen de la β-globina. En las formas autosómicas recesivas de la enfermedad, ambos padres deben ser portadores para que un niño resulte afectado. Si ambos padres son portadores de un rasgo de hemoglobinopatía, el riesgo es del 25 % por cada embarazo de un niño afectado. [27]
Los genes implicados en la talasemia controlan la producción de hemoglobina saludable. La hemoglobina fija el oxígeno en los pulmones y lo libera cuando los glóbulos rojos llegan a los tejidos periféricos, como el hígado. La fijación y liberación del oxígeno por parte de la hemoglobina son esenciales para la supervivencia. [ cita requerida ]
Evolución
Tener una única variante genética para la talasemia puede proteger contra la malaria y, por lo tanto, puede ser una ventaja. [28]
Normalmente, la mayor parte de la hemoglobina adulta ( HbA ) está compuesta por cuatro cadenas proteicas, dos cadenas de globina α y dos de globina β dispuestas en un heterotetrámero . En la talasemia, los pacientes tienen defectos en la región no codificante de los genes de globina α o β, lo que provoca una producción ineficaz de cadenas de globina alfa o beta normales, lo que puede conducir a una eritropoyesis ineficaz, destrucción prematura de glóbulos rojos y anemia. [8]
Las talasemias se clasifican según la cadena de la molécula de hemoglobina afectada. En las α-talasemias se afecta la producción de la cadena α-globina, mientras que en las β-talasemias se afecta la producción de la cadena β-globina. [30]
Las cadenas de β-globina están codificadas por un único gen en el cromosoma 11 ; las cadenas de α-globina están codificadas por dos genes estrechamente vinculados en el cromosoma 16. [ 31] Por lo tanto, en una persona sana con dos copias de cada cromosoma, dos loci codifican la cadena β y cuatro loci codifican la cadena α. La deleción de uno de los loci α tiene una alta prevalencia en personas de ascendencia africana o asiática, lo que las hace más propensas a desarrollar α-talasemia. Las β-talasemias no solo son comunes en africanos , sino también en griegos y turcos . [ cita requerida ]
Alfa-talasemias
Las α-talasemias afectan a los genes HBA1 [32] y HBA2 [33] , que se heredan de forma recesiva mendeliana . Existen dos loci genéticos y, por lo tanto, cuatro alelos. Existen dos loci genéticos para la α-globina, por lo que hay cuatro alelos en las células diploides. Dos alelos son de origen materno y dos alelos son de origen paterno. La gravedad de las α-talasemias se correlaciona con el número de α-globina afectados; cuanto mayor sea el número de alelos, más graves serán las manifestaciones de la enfermedad. [34] Las α-talasemias dan lugar a una disminución de la producción de α-globina; por lo tanto, se producen menos cadenas de α-globina, lo que da lugar a un exceso de cadenas β en adultos y un exceso de cadenas γ en recién nacidos. El exceso de cadenas β forma tetrámeros inestables (llamados hemoglobina H o HbH de 4 cadenas beta), que tienen curvas de disociación de oxígeno anormales. Las talasemias alfa se encuentran a menudo en personas del sudeste asiático, Oriente Medio, China y en personas de ascendencia africana. [35]
Beta-talasemia
Las beta talasemias se deben a mutaciones en el gen HBB del cromosoma 11 [36], que también se heredan de forma autosómica recesiva. La gravedad de la enfermedad depende de la naturaleza de la mutación y de la presencia de mutaciones en uno o ambos alelos. Los alelos mutados se denominan β + cuando la función parcial está conservada (o bien la proteína tiene una función reducida, o bien funciona normalmente pero se produce en cantidad reducida) o βo , cuando no se produce proteína funcional. La situación de ambos alelos determina el cuadro clínico:
La talasemia β mayor ( anemia mediterránea o anemia de Cooley ) es causada por un genotipo β o /β o . No se producen cadenas β funcionales y, por lo tanto, no se puede ensamblar la hemoglobina A. Esta es la forma más grave de talasemia β;
La talasemia β intermedia es causada por un genotipo β + /β o o β + /β + . En esta forma, se produce algo de hemoglobina A;
La talasemia β menor es causada por un genotipo β/β o o β/β + . Solo uno de los dos alelos de la β globina contiene una mutación, por lo que la producción de la cadena β no se ve gravemente comprometida y los pacientes pueden ser relativamente asintomáticos.
La beta talasemia se presenta con mayor frecuencia en personas de origen mediterráneo. En menor medida, también pueden verse afectadas personas de origen chino, asiático y afroamericano. [35]
Talasemia delta
Además de las cadenas alfa y beta presentes en la hemoglobina, aproximadamente el 3% de la hemoglobina adulta está formada por cadenas alfa y delta. Al igual que en el caso de la beta talasemia, pueden producirse mutaciones que afecten a la capacidad de este gen para producir cadenas delta. [37] [38]
Hemoglobinopatías combinadas
La talasemia puede coexistir con otras hemoglobinopatías . Las más frecuentes son:
Hemoglobina C /talasemia: común en poblaciones mediterráneas y africanas , la hemoglobina C/β o talasemia causa una anemia hemolítica moderadamente grave con esplenomegalia; la hemoglobina C/β + talasemia produce una enfermedad más leve. [ cita requerida ]
1) Hipogonadismo: la sobrecarga de hierro en las células gonadotrópicas de la hipófisis provoca una disminución de la secreción de gonadotropina, lo que conduce a una pubertad retrasada, lenta o detenida.
2) Hipoparatiroidismo: la anemia crónica provoca hematopoyesis que produce reabsorción ósea que disminuye la secreción paratiroidea.
3) Insuficiencia suprarrenal: disminución del crecimiento del vello púbico y axilar en pacientes adolescentes con TM. Esto se debe a un exceso de depósito de hierro que causa disfunción suprarrenal.
4) Hipotiroidismo: Aumento de peso y retraso del crecimiento en pacientes adolescentes. El secundario es poco frecuente y la mayoría padece hipotiroidismo primario.
[85]
Gestión
Dada la variedad de gravedades, algunas personas no requieren tratamiento (aquellas que son asintomáticas) mientras que otras requieren transfusiones de sangre regulares para sobrevivir. [43] Las personas con talasemia grave requieren tratamiento médico y el tratamiento principal suele ser una transfusión de glóbulos rojos.
Transfusiones de glóbulos rojos
Las transfusiones de sangre son el principal método de tratamiento para prolongar la vida. [44] El método y la frecuencia necesarios varían en cada persona según la gravedad, la edad, si la persona tiene retraso en el crecimiento, la presencia de eritropoyesis extramedular (pediatría), si una persona está embarazada y la salud cardíaca. Las transfusiones de sangre conllevan riesgos que incluyen empeoramiento de la sobrecarga de hierro, riesgo de infecciones, riesgo de formación de anticuerpos contra los glóbulos rojos, mayor riesgo de desarrollo de reacciones de hipersensibilidad y riesgo de inflamación de la vesícula biliar ( colecistitis ). [43]
Las transfusiones de sangre múltiples pueden provocar una sobrecarga de hierro. La sobrecarga de hierro relacionada con la talasemia puede tratarse mediante terapia de quelación con los medicamentos deferoxamina , deferiprona o deferasirox . [45] [46] [47] Estos tratamientos han dado como resultado una mayor esperanza de vida para las personas con talasemia mayor. [45] La deferoxamina solo es eficaz como inyección diaria, lo que complica su uso a largo plazo. Sin embargo, es económica y segura. Los efectos adversos incluyen reacciones cutáneas primarias alrededor del lugar de la inyección y pérdida de audición . [45] El deferasirox y la deferiprona son medicamentos orales, cuyos efectos secundarios comunes incluyen náuseas, vómitos y diarrea. Al comparar la efectividad, no hay evidencia de que el deferasirox o la deferiprona sean superiores, sin embargo, no se ha realizado una comparación a largo plazo. [47] El deferasirox no es eficaz para todos los pacientes y puede no ser adecuado para aquellos con problemas cardíacos importantes relacionados con la sobrecarga de hierro, mientras que la deferiprona parece ser el agente más eficaz cuando el corazón está afectado. Además, el costo del deferasirox también es significativo. [45] La combinación de medicamentos bloqueadores de los canales de calcio con terapia de quelación del hierro está en estudio, sin embargo, los beneficios no están claros a partir de los ensayos clínicos realizados. [48]
El trasplante de médula ósea puede ofrecer la posibilidad de curación en personas jóvenes que tienen un donante compatible con HLA . [50] Las tasas de éxito han alcanzado el rango del 80-90%. [50] La mortalidad por el procedimiento es de alrededor del 3%. [51] No existen ensayos controlados aleatorizados que hayan probado la seguridad y eficacia del trasplante de médula ósea de un donante no idéntico en personas con β-talasemia que dependen de transfusiones de sangre. [52]
Las enfermedades de injerto contra huésped (EICH) son un efecto secundario relevante del trasplante de médula ósea. Es necesario realizar más investigaciones para evaluar si las células estromales mesenquimales se pueden utilizar como profilaxis o tratamiento para la EICH. [53]
Si la persona no tiene un donante compatible con HLA, se puede intentar un trasplante de médula ósea de madre haploidéntica a hijo (donante no compatible). En un estudio de 31 personas, la tasa de supervivencia sin talasemia fue del 70%, la de rechazo del 23% y la de mortalidad del 7%. Los resultados más positivos tienden a darse en personas muy jóvenes. [54]
Terapia génica
Betibeglogene autotemcel , vendido bajo la marca Zynteglo, es una terapia génica para el tratamiento de la beta talasemia. [55] Fue aprobado para uso médico en la Unión Europea en mayo de 2019, [56] y en los Estados Unidos en agosto de 2022. [57] [58] Betibeglogene autotemcel está indicado para el tratamiento de personas de doce años o más con beta talasemia dependiente de transfusión que no tienen un genotipo β0/β0, para quienes el trasplante de células madre hematopoyéticas (HSC) es apropiado pero no hay un donante de HSC relacionado compatible con el antígeno leucocitario humano (HLA) disponible.
El procedimiento implica la recolección de células madre hematopoyéticas (CMH) de la sangre de la persona afectada. A continuación, se añade a las CMH un gen de beta-globina mediante un vector lentiviral . Después de destruir la médula ósea de la persona afectada con una dosis de quimioterapia (un régimen de acondicionamiento mieloablativo), las CMH alteradas se infunden de nuevo en la persona afectada, donde se injertan en la médula ósea, donde proliferan. Esto potencialmente da como resultado un aumento progresivo de la síntesis de hemoglobina A2 en todos los glóbulos rojos en desarrollo posteriores, con la consiguiente resolución de la anemia. [59]
El tratamiento fue aprobado en el Reino Unido para el tratamiento de la beta talasemia dependiente de transfusión en noviembre de 2023 [61] [62] [63] y en los Estados Unidos en enero de 2024. [64] [65] [66]
La terapia génica se realiza a partir de las propias células madre de la sangre del receptor, que se modifican y se administran como una infusión única de una sola dosis como parte de un trasplante de células madre hematopoyéticas . Antes del tratamiento, se recolectan las propias células madre del receptor y luego el receptor debe someterse a un acondicionamiento mieloablativo (quimioterapia de dosis alta), un proceso que extrae células de la médula ósea para que puedan reemplazarse con las células modificadas en exagamglogene autotemcel . Las células madre de la sangre modificadas se trasplantan nuevamente al receptor, donde se injertan dentro de la médula ósea y aumentan la producción de hemoglobina fetal (HbF), un tipo de hemoglobina que facilita el suministro de oxígeno.
Otros tratamientos
El tratamiento con hidroxiurea con el objetivo de reactivar los genes gamma para producir HbF no cuenta con evidencia de alta calidad que respalde su eficacia. [67]
No existen pruebas de ensayos controlados aleatorizados que respalden la administración de suplementos de zinc a pacientes con talasemia. [68] Se han sugerido programas informáticos o aplicaciones móviles como herramientas para ayudar a las personas a controlar la talasemia y seguir sus terapias, incluida la terapia de quelación del hierro. La eficacia de estas aplicaciones no se ha investigado en profundidad. [69]
Las personas con talasemia tienen un mayor riesgo de osteoporosis. [70] Las opciones de tratamiento incluyen bifosfonatos y, en ocasiones, la adición de terapia hormonal. Se han sugerido otros tratamientos, como calcitonina, zinc, hidroxiurea y suplementos de calcio. La eficacia de los bifosfonatos y el zinc no está clara y se requieren más estudios. [70]
Cuidado dental
Ayudar a las personas con talasemia con el cuidado dental y el tratamiento de los problemas dentales puede ser un desafío debido a la enfermedad subyacente. La sobrecarga de hierro debido a las transfusiones de sangre puede provocar la deposición de hierro en los dientes y la decoloración, y existe un mayor riesgo de infección. [71] Las opciones de tratamiento para las personas con talasemia deben modificarse para garantizar que se tengan en cuenta las necesidades de la persona y se recomienda encarecidamente la detección temprana y el manejo temprano de cualquier problema para reducir el riesgo de que la persona necesite tratamientos dentales más complicados. [71] La evidencia que respalda la efectividad de los tratamientos dentales en personas con talasemia es débil y se necesitan ensayos clínicos de mayor calidad. [71]
Talasemia leve
Las personas con rasgos de talasemia no requieren atención médica ni seguimiento después de que se realiza el diagnóstico inicial. [44] Se debe advertir a las personas con rasgos de β-talasemia que su condición puede ser diagnosticada erróneamente como la anemia por deficiencia de hierro más común . Deben evitar el uso rutinario de suplementos de hierro , pero la deficiencia de hierro puede desarrollarse durante el embarazo o por sangrado crónico. [72] El asesoramiento genético está indicado para todas las personas con trastornos genéticos, especialmente cuando la familia está en riesgo de una forma grave de la enfermedad que puede prevenirse. [73]
Prevención
El Colegio Estadounidense de Obstetras y Ginecólogos recomienda que todas las personas que estén pensando en quedarse embarazadas se hagan una prueba para ver si tienen talasemia. [74] Se recomienda asesoramiento genético y pruebas genéticas para las familias que tienen un rasgo de talasemia. [27] Conocer el riesgo genético, idealmente antes de formar una familia, permitiría a las familias comprender más sobre la enfermedad y tomar una decisión informada que sea mejor para su familia. [27]
En Chipre existe una política de detección para reducir la tasa de talasemia que, desde la implementación del programa en la década de 1970 (que también incluye detección prenatal y aborto), ha reducido el número de niños nacidos con la enfermedad de uno de cada 158 nacimientos a casi cero. [75] Grecia también tiene un programa de detección para identificar a las personas que son portadoras. [76]
En Irán , como prueba prematrimonial, primero se comprueban los índices de glóbulos rojos del hombre. Si tiene microcitosis ( hemoglobina corpuscular media < 27 pg o volumen corpuscular medio < 80 fl), se realiza la prueba a la mujer. Cuando ambos presentan microcitosis, se miden sus concentraciones de hemoglobina A2 . Si ambos tienen una concentración superior al 3,5% (diagnóstico de rasgo talasémico), se los deriva al puesto de salud local designado para recibir asesoramiento genético . [77]
En la India [78], tanto organizaciones gubernamentales como no gubernamentales están organizando campañas de concienciación a gran escala para promover la detección prematrimonial voluntaria y desaconsejar firmemente el matrimonio entre portadores.
Epidemiología
La forma beta de la talasemia es particularmente frecuente entre los pueblos mediterráneos , y esta asociación geográfica es responsable de su nombre original. [79] La talasemia provocó 25.000 muertes en 2013, en comparación con las 36.000 muertes en 1990. [15]
La enfermedad también se encuentra en poblaciones que viven en África, América y en la gente Tharu en la región Terai de Nepal y la India . [81] Se cree que explica tasas mucho más bajas de enfermedades y muertes por malaria, [82] lo que explica la capacidad histórica de los Tharus de sobrevivir en áreas con una fuerte infestación de malaria mientras que otros no pudieron. Las talasemias se asocian particularmente con personas de origen mediterráneo, árabes (especialmente palestinos y personas de ascendencia palestina) y asiáticos. [83] La prevalencia estimada es del 16% en personas de Chipre , 1% [84] en Tailandia y del 3 al 8% en poblaciones de Bangladesh , China , India , Malasia y Pakistán .
Se estima que aproximadamente el 1,5% de la población mundial (80 a 90 millones de personas) son portadores de β-talasemia. [85] Sin embargo, faltan datos exactos sobre las tasas de portadores en muchas poblaciones, en particular en las zonas en desarrollo del mundo que se sabe o se espera que estén muy afectadas. [86] [87] Debido a la prevalencia de la enfermedad en países con poco conocimiento de la talasemia, el acceso a un tratamiento y diagnóstico adecuados puede ser difícil. [88] Si bien existen algunas instalaciones de diagnóstico y tratamiento en los países en desarrollo, en la mayoría de los casos no son proporcionadas por los servicios gubernamentales y están disponibles solo para los pacientes que pueden pagarlas. En general, las poblaciones más pobres solo tienen acceso a instalaciones de diagnóstico y transfusiones de sangre limitadas. En algunos países en desarrollo, prácticamente no hay instalaciones para el diagnóstico o el tratamiento de la talasemia. [88]
Etimología y sinónimos
La palabra talasemia ( / θ æ l ɪ ˈ s iː m i ə / ) deriva del griego thalassa (θάλασσα), "mar", [89] y del neolatín -emia (de la raíz compuesta griega - aimia (-αιμία), de haima (αἷμα), "sangre"). [90] Fue acuñada porque la condición llamada "anemia mediterránea" fue descrita por primera vez en personas de etnias mediterráneas . La "anemia mediterránea" pasó a llamarse talasemia mayor una vez que se comprendieron mejor los genes. La palabra talasemia se utilizó por primera vez en 1932. [79] : 877 [91]
Investigación
Referencias
^ abcdefgh "¿Cuáles son los signos y síntomas de las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 16 de septiembre de 2016. Consultado el 5 de septiembre de 2016 .
^ abc "¿Qué causa las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 26 de agosto de 2016. Consultado el 5 de septiembre de 2016 .
^ ab "¿Cómo se diagnostican las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 16 de septiembre de 2016. Consultado el 5 de septiembre de 2016 .
^ abcde "¿Cómo se tratan las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 16 de septiembre de 2016. Consultado el 5 de septiembre de 2016 .
^ GBD 2015 Incidencia y prevalencia de enfermedades y lesiones (8 de octubre de 2016). "Incidencia, prevalencia y años vividos con discapacidad a nivel mundial, regional y nacional para 310 enfermedades y lesiones, 1990-2015: un análisis sistemático para el Estudio de la carga mundial de enfermedades 2015". Lancet . 388 (10053): 1545–1602. doi :10.1016/S0140-6736(16)31678-6. PMC 5055577 . PMID 27733282.{{cite journal}}: CS1 maint: nombres numéricos: lista de autores ( enlace )
^ ab GBD 2015 Mortalidad y causas de muerte (8 de octubre de 2016). "Esperanza de vida global, regional y nacional, mortalidad por todas las causas y mortalidad por causas específicas para 249 causas de muerte, 1980-2015: un análisis sistemático para el Estudio de la carga mundial de morbilidad 2015". Lancet . 388 (10053): 1459–1544. doi :10.1016/s0140-6736(16)31012-1. PMC 5388903 . PMID 27733281.{{cite journal}}: CS1 maint: nombres numéricos: lista de autores ( enlace )
^ abc "¿Qué son las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 26 de agosto de 2016. Consultado el 5 de septiembre de 2016 .
^ ab Baird DC, Batten SH, Sparks SK. Talasemia alfa y beta: revisión rápida de evidencia. Am Fam Physician. 1 de marzo de 2022;105(3):272-280. PMID 35289581.
^ "¿Cómo se pueden prevenir las talasemias?". NHLBI . 3 de julio de 2012. Archivado desde el original el 16 de septiembre de 2016. Consultado el 5 de septiembre de 2016 .
^ "Quelación de hierro" . Consultado el 15 de julio de 2020 .
^ Shah, Sandip; Sheth, Radhika; Shah, Kamlesh; Patel, Kinnari (febrero de 2020). "Seguridad y eficacia de la combinación de talidomida e hidroxiurea en la β-talasemia intermedia y mayor: un estudio piloto retrospectivo". British Journal of Haematology . 188 (3): e18–e21. doi : 10.1111/bjh.16272 . ISSN 0007-1048. PMID 31710694. S2CID 207940189.
^ Keikhaei, Bijan (2015). "Efectos clínicos y hematológicos de la hidroxiurea en pacientes con β-talasemia intermedia". Revista de investigación clínica y diagnóstica . 9 (10): OM01-3. doi :10.7860/JCDR/2015/14807.6660. PMC 4625280. PMID 26557561 .
^ Masera, Nicoletta; Tavecchia, Luisa; Capra, Marieta; Cazzaniga, Giovanni; Vimercati, Chiara; Pozzi, Lorena; Biondi, Andrea; Masera, Giuseppe (2010). "Respuesta óptima a la talidomida en un paciente con talasemia mayor resistente a la terapia convencional". Transfusión de sangre . 8 (1): 63–5. doi :10.2450/2009.0102-09. ISSN 1723-2007. PMC 2809513 . PMID 20104280.
^ Estudio de la carga mundial de enfermedades 2013 (22 de agosto de 2015). "Incidencia, prevalencia y años vividos con discapacidad a nivel mundial, regional y nacional para 301 enfermedades y lesiones agudas y crónicas en 188 países, 1990-2013: un análisis sistemático para el Estudio de la carga mundial de enfermedades 2013". Lancet . 386 (9995): 743–800. doi :10.1016/s0140-6736(15)60692-4. PMC 4561509 . PMID 26063472.{{cite journal}}: CS1 maint: nombres numéricos: lista de autores ( enlace )
^ ab GBD 2013 Mortalidad y causas de muerte (17 de diciembre de 2014). "Mortalidad global, regional y nacional específica por edad y sexo, por todas las causas y por causas específicas para 240 causas de muerte, 1990-2013: un análisis sistemático para el Estudio de la Carga Global de Enfermedades 2013". The Lancet . 385 (9963): 117–71. doi :10.1016/S0140-6736(14)61682-2. hdl :11655/15525. PMC 4340604 . PMID 25530442.{{cite journal}}: CS1 maint: nombres numéricos: lista de autores ( enlace )
^ Weatherall, DJ (2015). "Las talasemias: trastornos de la síntesis de globina". Williams Hematology (novena edición). McGraw Hill Professional. pág. 725. ISBN9780071833011.
^ Cianciulli P (octubre de 2008). "Tratamiento de la sobrecarga de hierro en la talasemia". Pediatr Endocrinol Rev. 6 ( Supl. 1): 208–13. PMID 19337180.
^ "Talasemia: síntomas y causas". Mayo Clinic . Archivado desde el original el 20 de noviembre de 2016. Consultado el 4 de abril de 2017 .
^ Vogiatzi, Maria G; Macklin, Eric A; Fung, Ellen B; Cheung, Angela M; Vichinsky, Elliot; Olivieri, Nancy; Kirby, Melanie; Kwiatkowski, Janet L; Cunningham, Melody; Holm, Ingrid A; Lane, Joseph; Schneider, Robert; Fleisher, Martin; Grady, Robert W; Peterson, Charles C; Giardina, Patricia J (marzo de 2009). "Enfermedad ósea en la talasemia: un problema frecuente y aún sin resolver". Revista de investigación ósea y mineral . 24 (3): 543–557. doi :10.1359/jbmr.080505. ISSN 0884-0431. PMC 3276604 . PMID 18505376.
^ "Síntomas y causas: agrandamiento del bazo (esplenomegalia) – Mayo Clinic". www.mayoclinic.org . Archivado desde el original el 19 de noviembre de 2016 . Consultado el 2 de febrero de 2017 .
^ Soliman, Ashraf T; Kalra, Sanjay; De Sanctis, Vincenzo (1 de noviembre de 2014). "Anemia y crecimiento". Revista India de Endocrinología y Metabolismo . 18 (7): S1–5. doi : 10.4103/2230-8210.145038 . PMC 4266864 . PMID 25538873.
^ "Complicaciones de la talasemia". Talasemia . Publicación abierta. Archivado desde el original el 3 de octubre de 2011 . Consultado el 27 de septiembre de 2011 .
^ Weatherall DJ. La nueva genética y la práctica clínica, Oxford University Press, Oxford 1991.
^ Huisman TH. Estructura y función de las hemoglobinas normales y anormales. En: Baillière's Clinical Haematology, Higgs DR, Weatherall DJ (Eds), WB Saunders, Londres 1993. p.1.
^ Natarajan K, Townes TM, Kutlar A. Trastornos de la estructura de la hemoglobina: anemia de células falciformes y anomalías relacionadas. En: Williams Hematology, 8.ª ed., Kaushansky K, Lichtman MA, Beutler E, et al. (Eds.), McGraw-Hill, 2010. pág. 48.
^ Wai Feng Lim, Logeswaran Muniandi, Elizabeth George, Jameela Sathar, Lai Kuan Teh y Mei I Lai (2015) HbF en HbE/β-talasemia: una correlación clínica y de laboratorio, Hematología, 20:6, 349-353, DOI: 10.1179/1607845414Y.0000000203
^ abc Hussein, Norita; Henneman, Lidewij; Kai, Joe; Qureshi, Nadeem (11 de octubre de 2021). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Evaluación del riesgo preconcepcional para talasemia, anemia de células falciformes, fibrosis quística y enfermedad de Tay-Sachs". Base de Datos Cochrane de Revisiones Sistemáticas . 2021 (10): CD010849. doi :10.1002/14651858.CD010849.pub4. PMC 8504980. PMID 34634131.
^ Wambua S; Mwangi, Tabitha W.; Kortok, Moses; Uyoga, Sophie M.; Macharia, Alex W.; Mwacharo, Jedidah K.; Weatherall, David J.; Snow, Robert W.; Marsh, Kevin; Williams, Thomas N. (mayo de 2006). "El efecto de la α+-talasemia en la incidencia de la malaria y otras enfermedades en niños que viven en la costa de Kenia". PLOS Medicine . 3 (5): e158. doi : 10.1371/journal.pmed.0030158 . PMC 1435778 . PMID 16605300.
^ Tassiopoulos S; Defteros, Spyros; Konstantopoulos, Kostas; Farmakis, Dimitris; Tsironi, María; Kyriakidis, Michalis; Esosopos, Atanasio (2005). "¿La beta-talasemia heterocigótica confiere protección contra la enfermedad de las arterias coronarias?". Anales de la Academia de Ciencias de Nueva York . 1054 (1): 467–70. Código Bib : 2005NYASA1054..467T. doi : 10.1196/anales.1345.068. PMID 16339699. S2CID 71993591.
^ Herbert l. Muncie, Jr; Campbell, James S. (15 de agosto de 2009). "Talasemia alfa y beta". American Family Physician . 80 (4): 339–344. PMID 19678601.
^ ab Galanello, Renzo; Cao, Antonio (5 de enero de 2011). "Alfa-talasemia". Genética en Medicina . 13 (2): 83–88. doi : 10.1097/GIM.0b013e3181fcb468 . ISSN 1098-3600. PMID 21381239.
^ ab "Los conceptos básicos de la anemia". WebMD . Consultado el 9 de mayo de 2019 .
^ "Delta-beta-talasemia". Orphanet . Consultado el 16 de septiembre de 2016 .
^ "HBD - subunidad delta de la hemoglobina". Orphanet . Consultado el 17 de septiembre de 2016 .
^ Torres Lde S (marzo de 2015). "Hemoglobina D-Punjab: origen, distribución y diagnóstico de laboratorio". Revista Brasileira de Hematología y Hemoterapia . 37 (2): 120-126. doi :10.1016/j.bjhh.2015.02.007. PMC 4382585 . PMID 25818823.
^ "¿Cómo se diagnostican las talasemias? – NHLBI, NIH". www.nhlbi.nih.gov . Archivado desde el original el 28 de julio de 2017 . Consultado el 6 de septiembre de 2017 .
^ Keohane, E; Smith, L; Walenga, J (2015). Hematología de Rodak: principios clínicos y aplicaciones (5.ª ed.). Elsevier Health Sciences. págs. 467–9. ISBN978-0-323-23906-6.
^ Kottke-Marchant, K; Davis, B (2012). Práctica de hematología de laboratorio (1.ª edición). John Wiley & Sons. pág. 569. ISBN978-1-4443-9857-1.
^ ab Foong, Wai Cheng; Loh, C Khai; Ho, Jacqueline J; Lau, Doris SC (13 de enero de 2023). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Inductores de hemoglobina fetal para reducir la transfusión sanguínea en beta-talasemias no dependientes de transfusión". Base de Datos Cochrane de Revisiones Sistemáticas . 2023 (1): CD013767. doi :10.1002/14651858.CD013767.pub2. PMC 9837847. PMID 36637054 .
^ ab Talasemia pediátrica ~ tratamiento en eMedicine
^ abcd Neufeld, EJ (2010). "Actualización sobre quelantes de hierro en la talasemia". Hematología . 2010 : 451–5. doi : 10.1182/asheducation-2010.1.451 . PMID 21239834.
^ Fisher, Sheila A.; Brunskill, Susan J.; Doree, Carolyn; Chowdhury, Onima; Gooding, Sarah; Roberts, David J. (21 de agosto de 2013). "Deferiprona oral para la quelación del hierro en personas con talasemia". Base de Datos Cochrane de Revisiones Sistemáticas (8): CD004839. doi :10.1002/14651858.CD004839.pub3. ISSN 1469-493X. PMID 23966105.
^ ab Bollig, Claudia; Schell, Lisa K.; Rücker, Gerta; Allert, Roman; Motschall, Edith; Niemeyer, Charlotte M.; Bassler, Dirk; Meerpohl, Joerg J. (15 de agosto de 2017). "Deferasirox para el manejo de la sobrecarga de hierro en personas con talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 2017 (8): CD007476. doi :10.1002/14651858.CD007476.pub3. ISSN 1469-493X. PMC 6483623. PMID 28809446 .
^ Sadaf, Alina; Hasan, Babar; Das, Jai K; Colan, Steven; Alvi, Najveen (12 de julio de 2018). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Bloqueadores de los canales de calcio para la prevención de la miocardiopatía debida a la sobrecarga de hierro en personas con beta talasemia dependiente de transfusiones". Base de Datos Cochrane de Revisiones Sistemáticas . 2018 (7): CD011626. doi :10.1002/14651858.CD011626.pub2. PMC 6513605. PMID 29998494 .
^ Ngim, CF; Lai, NM; Hong, JY; Tan, SL; Ramadas, A; Muthukumarasamy, P; Thong, MK (28 de mayo de 2020). "Terapia con hormona de crecimiento para personas con talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 2020 (5): CD012284. doi :10.1002/14651858.CD012284.pub3. PMC 7387677. PMID 32463488 .
^ ab Gaziev, J; Lucarelli, G (junio de 2011). "Trasplante de células madre hematopoyéticas para la talasemia". Investigación y terapia actuales con células madre . 6 (2): 162–9. doi :10.2174/157488811795495413. PMID 21190532.
^ Sabloff, M; Chandy, M; Wang, Z; Logan, BR; Ghavamzadeh, A; Li, CK; Irfan, SM; Bredeson, CN; et al. (2011). "Trasplante de médula ósea entre hermanos con HLA compatible para β-talasemia mayor". Blood . 117 (5): 1745–50. doi :10.1182/blood-2010-09-306829. PMC 3056598 . PMID 21119108.
^ Sharma, Akshay; Jagannath, Vanitha A.; Puri, Latika (21 de abril de 2021). "Trasplante de células madre hematopoyéticas para personas con β-talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 2021 (4): CD008708. doi :10.1002/14651858.CD008708.pub5. ISSN 1469-493X. PMC 8078520. PMID 33880750 .
^ Fisher, Sheila A; Cutler, Antony; Doree, Carolyn; Brunskill, Susan J; Stanworth, Simon J; Navarrete, Cristina; Girdlestone, John (30 de enero de 2019). Grupo Cochrane de Neoplasias Hematológicas (ed.). "Células estromales mesenquimales como tratamiento o profilaxis para la enfermedad de injerto contra huésped aguda o crónica en receptores de trasplante de células madre hematopoyéticas (TCMH) con una afección hematológica". Base de Datos Cochrane de Revisiones Sistemáticas . 1 (1): CD009768. doi :10.1002/14651858.CD009768.pub2. PMC 6353308 . PMID 30697701.
^ Sodani, P; Isgró, A; Gaziev, J; Paciaroni, K; Marziali, M; Simone, MD; Roveda, A; De Angelis, G; et al. (2011). "Trasplante de células madre hla-haploidénticas empobrecidas en células T en pacientes jóvenes con talasemia". Informes pediátricos . 3 (Suplemento 2): e13. doi :10.4081/pr.2011.s2.e13. PMC 3206538 . PMID 22053275.
^ Negro, Olivier; Eggimann, Anne-Virginie; Beuzard, Yves; Ribeil, Jean-Antoine; Bourget, Philippe; Borwornpinyo, Suparerk; Hongeng, Suradej; Hacein-Bey, Salima; Cavazzana, Marina; Leboulch, Philippe; Payen, Emmanuel (febrero de 2016). "Terapia génica de las β-hemoglobinopatías mediante transferencia lentiviral del gen β". Terapia genética humana . 27 (2): 148–165. doi :10.1089/hum.2016.007. PMC 4779296 . PMID 26886832.
^ «Zynteglo EPAR». Agencia Europea de Medicamentos (EMA) . 25 de marzo de 2019. Archivado desde el original el 16 de agosto de 2019. Consultado el 16 de agosto de 2019 .El texto se ha copiado de esta fuente, cuyos derechos de autor pertenecen a la Agencia Europea de Medicamentos. Se autoriza la reproducción siempre que se cite la fuente.
^ "Zynteglo". Administración de Alimentos y Medicamentos de Estados Unidos . 17 de agosto de 2022. Archivado desde el original el 26 de agosto de 2022. Consultado el 26 de agosto de 2022 .
^ "La FDA aprueba la primera terapia génica basada en células para tratar a pacientes adultos y pediátricos con beta-talasemia que requieren transfusiones de sangre periódicas". Administración de Alimentos y Medicamentos de Estados Unidos (FDA) (Comunicado de prensa). 17 de agosto de 2022. Archivado desde el original el 21 de agosto de 2022 . Consultado el 20 de agosto de 2022 .Este artículo incorpora texto de esta fuente, que se encuentra en el dominio público .
^ Biffi, A (19 de abril de 2018). "La terapia génica como opción curativa para la β-talasemia". The New England Journal of Medicine . 378 (16): 1551–1552. doi :10.1056/NEJMe1802169. PMID 29669229.
^ Stein R (31 de octubre de 2023). "Los asesores de la FDA no ven obstáculos para el tratamiento de la anemia falciforme con edición genética". NPR . Archivado desde el original el 4 de diciembre de 2023 . Consultado el 4 de diciembre de 2023 .
^ "La MHRA autoriza la primera terapia génica del mundo que tiene como objetivo curar la anemia falciforme y la beta-talasemia dependiente de transfusiones". Agencia Reguladora de Medicamentos y Productos Sanitarios (MHRA) (Comunicado de prensa). 16 de noviembre de 2023. Archivado desde el original el 25 de noviembre de 2023 . Consultado el 8 de diciembre de 2023 .
^ Sheridan C (noviembre de 2023). «Se aprueba la primera terapia CRISPR del mundo: ¿quién la recibirá?». Nature Biotechnology . 42 (1): 3–4. doi :10.1038/d41587-023-00016-6. PMID: 37989785. S2CID : 265350318. Archivado desde el original el 4 de diciembre de 2023. Consultado el 4 de diciembre de 2023 .
^ "Vertex y CRISPR Therapeutics anuncian la autorización de la primera terapia de edición genética CRISPR/Cas9, Casgevy (exagamglogene autotemcel), por parte de la MHRA del Reino Unido para el tratamiento de la anemia de células falciformes y la beta talasemia dependiente de transfusiones" (Comunicado de prensa). Vertex Pharmaceuticals. 16 de noviembre de 2023. Archivado desde el original el 22 de noviembre de 2023. Consultado el 9 de diciembre de 2023 a través de Business Wire.
^ ab "La FDA aprueba las primeras terapias genéticas para tratar a pacientes con anemia falciforme". Administración de Alimentos y Medicamentos de Estados Unidos (FDA) . 8 de diciembre de 2023. Archivado desde el original el 8 de diciembre de 2023. Consultado el 8 de diciembre de 2023 .Este artículo incorpora texto de esta fuente, que se encuentra en el dominio público .
^ "Vertex y CRISPR Therapeutics anuncian la aprobación por parte de la FDA de EE. UU. de Casgevy (exagamglogene autotemcel) para el tratamiento de la anemia de células falciformes" (Comunicado de prensa). Vertex Pharmaceuticals. 8 de diciembre de 2023. Archivado desde el original el 9 de diciembre de 2023. Consultado el 9 de diciembre de 2023 a través de Business Wire.
^ Comisionado, Oficina del (16 de enero de 2024). «FDA Roundup: January 16, 2024» (Resumen de la FDA: 16 de enero de 2024). FDA . Consultado el 19 de enero de 2024 .
^ Ansari, Saqib H; Lassi, Zohra S; Khowaja, Salima M; Adil, Syed Omair; Shamsi, Tahir S (16 de marzo de 2019). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Hidroxiurea (hidroxicarbamida) para la β-talasemia dependiente de transfusión". Base de Datos Cochrane de Revisiones Sistemáticas . 3 (3): CD012064. doi :10.1002/14651858.CD012064.pub2. PMC 6421980 . PMID 30882896.
^ Kye Mon Min Swe (2013). "Suplementos de zinc para el tratamiento de la talasemia y la anemia falciforme". Base de Datos Cochrane de Revisiones Sistemáticas . 2013 (6): CD009415. doi :10.1002/14651858.CD009415.pub2. PMC 9964104. PMID 23807756 .
^ Mulimani, Priti; Abas, Adinegara BL; Karanth, Laxminarayan; Colombatti, Raffaella; Kulkarni, Palna (2 de agosto de 2019). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Tratamiento de las complicaciones dentales y ortodóncicas en la talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 8 (8): CD012969. doi :10.1002/14651858.CD012969.pub2. PMC 6699676 . PMID 31425614.
^ ab Bhardwaj, Amit; Swe, Kye Mon Min; Sinha, Nirmal K.; Osunkwo, Ifeyinwa (10 de marzo de 2016). "Tratamiento para la osteoporosis en personas con ß-talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 3 : CD010429. doi :10.1002/14651858.CD010429.pub2. ISSN 1469-493X. PMID 26964506.
^ abc Mulimani, Priti; Abas, Adinegara BL; Karanth, Laxminarayan; Colombatti, Raffaella; Kulkarni, Palna (2 de febrero de 2023). Grupo Cochrane de Fibrosis Quística y Trastornos Genéticos (ed.). "Tratamiento de las complicaciones dentales y ortodóncicas en la talasemia". Base de Datos Cochrane de Revisiones Sistemáticas . 2023 (2): CD012969. doi :10.1002/14651858.CD012969.pub3. PMC 9893875. PMID 36732291 .
^ Burdick CO; Ntaios, G.; Rathod, D. (marzo de 2009). "Separación del rasgo de talasemia y la deficiencia de hierro mediante una simple inspección". Am. J. Clin. Pathol . 131 (3): 444, respuesta del autor 445. doi : 10.1309/AJCPC09VRAXEASMH . PMID 19228649.
^ "Detección de portadores en la era de la medicina genómica – ACOG". www.acog.org . Archivado desde el original el 25 de febrero de 2017 . Consultado el 24 de febrero de 2017 .
^ Leung TN; Lau TK; Chung TKh (abril de 2005). "Detección de talasemia durante el embarazo". Current Opinion in Obstetrics and Gynecology . 17 (2): 129–34. doi :10.1097/01.gco.0000162180.22984.a3. PMID 15758603. S2CID 41877258.
^ Loukopoulos, D (octubre de 2011). "Hemoglobinopatías en Grecia: programa de prevención durante los últimos 35 años". The Indian Journal of Medical Research . 134 (4): 572–6. PMC 3237258 . PMID 22089622.
^ Samavat A, Modell B (noviembre de 2004). "Programa nacional iraní de detección de talasemia". BMJ (Clinical Research Ed.) . 329 (7475): 1134–7. doi :10.1136/bmj.329.7475.1134. PMC 527686. PMID 15539666 .
^ Petrou, Mary (1 de enero de 2010). "Detección de beta talasemia". Indian Journal of Human Genetics . 16 (1): 1–5. doi : 10.4103/0971-6866.64934 . PMC 2927788 . PMID 20838484.[ enlace muerto permanente ]
^ ab Greer, John P.; Arber, Daniel A.; Glader, Bertil; List, Alan F.; Means, Jr., Robert T.; Paraskevas, Frixos; Rodgers, George M.; Foerster, John (2013). Hematología clínica de Wintrobe . Wolters Kluwer, Lippincott Williams & Wilkins Health. ISBN9781451172683.
^ Waheed, Fazeela; Fishter, Colleen; Awofeso, AwoNiyi; Stanley, David (julio de 2016). "Detección de portadores de beta-talasemia en las Maldivas: percepciones de los padres de niños afectados que no participaron en la detección y sus consecuencias". Journal of Community Genetics . 7 (3): 243–253. doi :10.1007/s12687-016-0273-5. PMC 4960032 . PMID 27393346.
^ Modiano, G.; Morpurgo, G.; Terrenato, L.; Novelletto, A.; Di Rienzo, A.; Colombo, B.; Purpura, M.; Mariani, M.; et al. (1991). "Protección contra la morbilidad por malaria: casi fijación del gen de la α-talasemia en una población nepalí". American Journal of Human Genetics . 48 (2): 390–7. PMC 1683029 . PMID 1990845.
^ Terrenato, L; Shrestha, S; Dixit, KA; Luzzatto, L; Modiano, G; Morpurgo, G; Arese, P (febrero de 1988). "Disminución de la morbilidad por malaria en el pueblo Tharu en comparación con las poblaciones simpátricas en Nepal". Anales de Medicina Tropical y Parasitología . 82 (1): 1–11. doi :10.1080/00034983.1988.11812202. PMID 3041928.
^ "Talasemia" (en tailandés). Departamento de Ciencias Médicas. Septiembre de 2011. Archivado desde el original el 25 de septiembre de 2011.
^ Galanello, Renzo; Origa, Raffaella (2010). "Beta-talasemia". Revista Orphanet de Enfermedades Raras . 5 (1): 11. doi : 10.1186/1750-1172-5-11 . PMC 2893117 . PMID 20492708.
^ Galanello, Renzo; Origa, Raffaella (2010). "Beta-talasemia". Revista Orphanet de Enfermedades Raras . 5 (1): 11. doi : 10.1186/1750-1172-5-11 . PMC 2893117 . PMID 20492708.
^ Vichinsky, Elliott P. (1 de noviembre de 2005). "Patrones cambiantes de la talasemia en todo el mundo". Anales de la Academia de Ciencias de Nueva York . 1054 (1): 18–24. Bibcode :2005NYASA1054...18V. doi :10.1196/annals.1345.003. ISSN 1749-6632. PMID 16339647. S2CID 26329509.
^ ab Weatherall, David J. (noviembre de 2005). "Discurso inaugural: El desafío de la talasemia para los países en desarrollo". Anales de la Academia de Ciencias de Nueva York . 1054 (1): 11–17. Bibcode :2005NYASA1054...11W. doi :10.1196/annals.1345.002. PMID 16339646. S2CID 45770891.
^ Whipple, GH; Bradford, WI (1932). "Anemia racial o familiar en niños asociada con trastornos fundamentales del metabolismo óseo y pigmentario (Cooley-Von Jaksch)". American Journal of Diseases of Children . 44 : 336–365. doi :10.1001/archpedi.1932.01950090074009.
De Sanctis, Vincenzo; Soliman, Ashraf T. 1 ; Elsedfy, Heba 2 ; Skordis, Nicos 3 ; Kattamis, Cristos 4 ; Angastiniotis, Michael 5 ; Karimi, Mehran 6 ; Yassin, Mohd Abdel Daem Mohd 7 ; El Awwa, Ahmed 1 ; Stoeva, Ivan 8 ; Raiola, Giuseppe 9 ; Galati, María Concetta 10 ; Bedair, Elsaid M. 11 ; Fiscina, Bernadette 12 ; El Kholy, Mohamed 2 Información del autor Indian Journal of Endocrinology and Metabolism 17(1):p 8-18, enero-febrero de 2013. | DOI: 10.4103/2230-8210.107808
Enlaces externos
Aprendiendo sobre la talasemia publicado por el Instituto Nacional de Investigación del Genoma Humano.