Tecnologías de transcriptómica
Entonces, el ARNm sirve como una molécula intermediaria y transitoria en la red de información, mientras que los ARNs no codificantes cumplen con diferentes funciones adicionales.
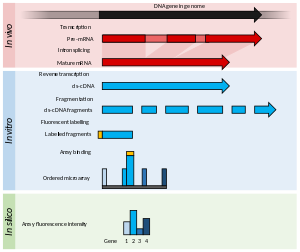
Las tecnologías de transcriptómica permiten saber qué procesos celulares se encuentran activos y cuáles inactivos.
Existen dos técnicas actuales clave en este campo: microarrays, los cuales cuantifican un conjunto de secuencias predeterminadas, y ARN-Seq, el cual utiliza secuenciación de alto rendimiento para registrar todos los transcritos.
Sin el conocimiento de experimentos previos, sería imposible interpretar la información contenida en un transcriptoma.
También se puede utilizar para inferir las funciones de genes previamente no anotados.
[6][7][8] El rápido desarrollo de nuevas tecnologías con sensibilidad mejorada y más baratas ha posibilitado esta explosión en transcriptómica.
[17][18] Sin embargo, estos métodos son laboriosos y solo pueden capturar una pequeña subsección del transcriptoma.
[39] El primer proyecto de ARN-Seq se publicó en 2006 con 1000 transcritos secuenciados utilizando tecnología 454.
[44][45] El ARN aislado puede ser tratado adicionalmente con DNasa para digerir cualquier traza de ADN.
Es típica la congelación rápida de tejidos previa al aislamiento del ARN.
Así se reduce la exposición de las enzimas RNasa una vez que el aislamiento está completo.
En la actualidad, los transcriptomas de célula única están bien descritos e incluso se han extendido a ARN-Seq in situ, en el que los transcriptomas de células individuales se analizan directamente en tejidos fijados.
Sin embargo, la ribo-depleción también puede introducir cierto sesgo al eliminar de manera inespecífica transcritos fuera del objetivo.
[67] La fragmentación se puede lograr mediante hidrólisis química, nebulización, sonicación o transcripción inversa con nucleótidos terminadores de cadena.
[71] Los identificadores moleculares únicos (UMIs) son secuencias cortas al azar que se utilizan para marcar individualmente fragmentos de secuencia durante la preparación de genotecas para que cada fragmento marcado sea único.
Los datos de secuenciación se pueden almacenar en repositorios públicos, tales como el Sequence Read Archive (SRA).
Adicionalmente, se deben identificar los artefactos en imágenes y eliminarlos del análisis general.
El proceso se puede dividir en cuatro etapas: control de calidad, alineamiento, cuantificación y expresión diferencial.
[105][106] Las anormalidades se pueden eliminar o marcar para tratamientos especiales en procesos posteriores.
El software kallisto es un método que combina el pseudoalineamiento y cuantificación en un solo paso, ejecutándose 2 órdenes de magnitud más rápido que otros métodos contemporáneos, tales como aquellos utilizados por tophat/cufflinks, con menos carga computacional.
Los análisis de transcriptómica se pueden validar utilizando una técnica independiente, por ejemplo, una PCR cuantitativa (qPCR), la cual es reconocible y estadísticamente evaluable.
La medición por qPCR es similar a la obtenida por ARN-Seq, en la que se puede calcular un valor para la concentración de una región diana en una muestra dada.
[62][137][138] La validación funcional de genes clave es una consideración importante para la planificación posterior a la construcción del transcriptoma.
[144][145] El ARN-Seq de patógenos humanos se ha convertido en un método establecido para cuantificar cambios en la expresión génica, identificando nuevos factores de virulencia, prediciendo resistencia a antibióticos, y desentrañando interacciones inmunitarias huésped-patógeno.
[145] El análisis transcriptómico se ha centrado predominantemente en ya sea el huésped o el patógeno.
Esta técnica permite estudiar la respuesta dinámica y redes reguladoras de genes inter-especie en ambas partes involucradas en la interacción desde el contacto inicial hasta la invasión y la persistencia final del patógeno o su eliminación por el sistema inmunitario del huésped.
[151] Los perfiles transcriptómicos también aportan información crucial sobre los mecanismos de resistencia a fármacos.
[159] El ARN-Seq también se puede utilizar para identificar regiones codificantes de proteínas previamente desconocidas en genomas ya secuenciados.
Sin embargo, una aproximación que utiliza el escalado temporal y binarización de transcriptomas para definir un conjunto de genes que predice la edad biológica con precisión permitió alcanzar una evaluación cercana al límite teórico.
[160] La transcriptómica se aplican más comúnmente al contenido en ARNm de una células.