Reacción de Stille
Se ha sugerido que los ligandos aniónicos, como OAc, aceleran este paso mediante la formación de [Pd (OAc) (PR3) n] -, haciendo que la especie de paladio sea más nucleófila.
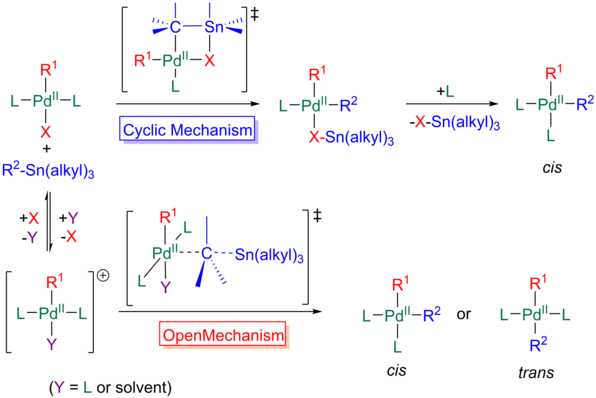
Se cree que esto ocurre a través de dos mecanismos.
Otro mecanismo comúnmente visto implica la misma adición inicial del organostanano al complejo de paladio trans como se vio anteriormente; sin embargo, en este caso, el grupo X no se coordina con el estaño, produciendo un estado de transición abierto.
[30] Una vía menos común para la transmetalación es a través de un mecanismo disociativo o asistido por solvente.
Por lo tanto, cualquier transducto debe isomerizarse en el intermedio cis o el acoplamiento se frustrará.
Existe una variedad de mecanismos para la eliminación reductiva y estos generalmente se consideran concertados.
Esta reacción ocurre en dos pasos: primero, la eliminación reductiva es seguida por la coordinación del enlace sigma recién formado entre R1 y R2 con el metal, con la disociación final que produce el producto acoplado.
[11][31][32] Finalmente, un ligando adicional puede asociarse al paladio para formar una estructura bipiramidal trigonal de 18 electrones, con R1 y R2 cis entre sí en posiciones ecuatoriales.
[35] Normalmente, se utilizan ligandos de donación intermedia, como las fosfinas.
[36] El aditivo más común a la reacción de Stille es el cobre cocatalítico (I), específicamente el yoduro cuproso, que puede aumentar las tasas hasta> 103 veces.
Al igual que en el mecanismo cíclico, un ligando neutro, como la fosfina, debe disociarse en lugar del grupo aniónico X.
Se cree que procede a través de dos posibles mecanismos.
En segundo lugar, el catalizador de Pd (0) puede experimentar un proceso radical para producir el dímero.
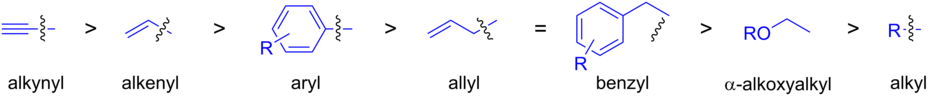
Como se vio anteriormente, los grupos alquilo son normalmente los más lentos en migrar al catalizador de paladio.
[10] También se ha encontrado que a temperaturas tan bajas como 50 °C, los grupos arilo en el paladio y una fosfina coordinada pueden intercambiar.
Aunque normalmente no se detectan, pueden ser un posible producto secundario en muchos casos.
[10] Finalmente, una reacción secundaria bastante rara y exótica se conoce como sustitución cine.
Después de la eliminación del hidruro β, la inserción migratoria y la protodestanilización, se puede sintetizar una olefina 1,2-disustituida.
[10] Pueden ocurrir otras numerosas reacciones secundarias, y estas incluyen la isomerización E / Z, que potencialmente puede ser un problema cuando se utiliza un alquenilstanano.
Normalmente, los organostananos son bastante estables a la hidrólisis, sin embargo, cuando se usan aril stannanos muy ricos en electrones, esto puede convertirse en una reacción secundaria significativa.
Los heterociclos sustituidos con halógeno también se han utilizado como parejas de acoplamiento, incluidas piridinas, furanos, tiofenos, tiazoles, indoles, imidazoles, purinas, uracilo, citosinas, pirimidinas y más (consulte a continuación la tabla de heterociclos; los halógenos se pueden sustituir en una variedad de posiciones en cada uno).
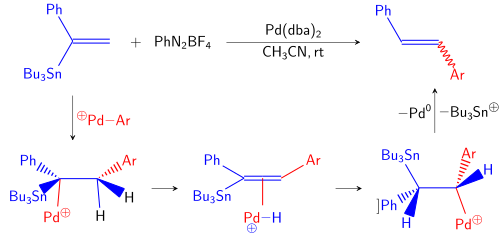
Mientras se emplean comúnmente, los haluros alílicos proceden a través de un estado de transición η3, lo que permite el acoplamiento con el organostanano en la posición α o γ, que ocurre predominantemente en el carbono menos sustituido (ver ejemplo a continuación).
Los reactivos de organoestaño son estables al aire y a la humedad.
Por ejemplo, en el siguiente caso, el vinilstanano sustituido con α solo reacciona con un yoduro terminal debido al impedimento estérico.
La única limitación para estos reactivos son los sustituyentes en la posición orto tan pequeños como los grupos metilo pueden disminuir la velocidad de reacción.
[10][49] Los alquinilstannanos, los stannanes más reactivos, también se han utilizado en acoplamientos Stille.
Los reactivos de distannane y acyl stannane también se han utilizado en acoplamientos Stille.
El organostanano complejo está acoplado a dos grupos yoduro de arilo.
Aquí, el organostanano tiene dos grupos terminales de tributil estaño atacados a un alqueno.
exploró cómo podría usarse el acoplamiento cruzado de Stille-carbonilativo para sintetizar benzofenonas.

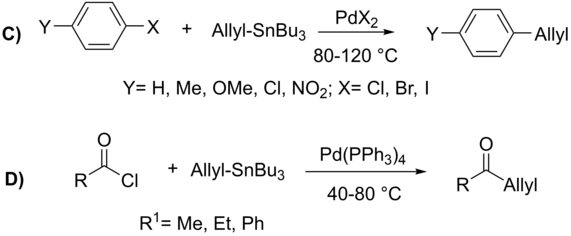



























![Síntesis de benzo[4,5]furopyridines](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/879px-Benzofuropyridines.png)