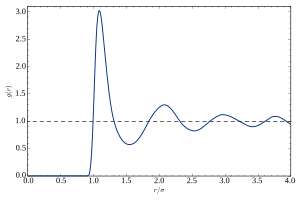
Si se toma una partícula dada como ubicada en el origen O, y si ρ = N/V es la densidad numérica promedio, entonces la densidad local promediada en el tiempo a una distancia r desde O es ρg(r).Esta definición simplificada es válida para un sistema isotrópico y homogéneo.La función de distribución radial se determina usualmente calculando la distancia entre todos los pares de partículas y colocando los datos en un histograma.Entonces, el histograma se normaliza con respecto a un gas ideal completamente descorrelacionado.En tres dimensiones, esta normalización es la densidad numérica del sistema, multiplicada por el volumen del cascarón esférico; es decir: donde ρ es la densidad numérica.Considérese un sistema de N partículas en un volumen V (para una densidad numérica promedio ρ = N/V) a una temperatura T. Definamos también el parámetro β = 1/(kT), donde k es la constante de Boltzmann.Las coordenadas de las partículas son ri , con i = 1,…,N.La energía potencial debida a la interacción entre las partículas es UN(r1,…,rN) y no consideramos el caso de un campo aplicado externamente.Los promedios apropiados se toman en el ensamble canónico (N,V,T) con la integral configuracional sobre todos las posibles combinaciones de posiciones de partículas: La probabilidad de encontrar a la partícula 1 en el intervalo dr1, a la partícula 2 en dr2, etc. está dada por (1)Dado que el número de partículas es muy grande, P(N) no es muy útil por sí mismo.No obstante, se puede obtener al probabilidad de una configuración reducida, donde las posiciones de únicamente n < N partículas están fijas en r1,…,rn , sin restricciones sobre las N−n partículas restantes.Hasta este punto, es necesario integrar (1) sobre las coordenadas restantes rn+1,…,rN : Ya que las partículas son idénticas, es más relevante considerar la probabilidad de que cualesquiera n de ellas ocupen las posiciones r1,…,rn , en cualquier permutación, definiendo así la densidad de n partículas: (2)Para n = 1, (2) da la densidad de 1 partícula, la cual, para un cristal es una función periódica con máximos puntiagudos en los sitios de la red.La función g(n) es conocida como función de correlación, dado que, si los átomos son independientes unos de otros, ρ(n) sería simplemente igual a ρn.Si tomamos a la partícula 0 como fija en el origen de coordenadas, ρg(r) dr = dn(r) es el número de partículas (entre las N−1 restantes) que se encuentran en el volumen dr alrededor de la posición r. Formalmente, podemos contar estas partículas como donde los paréntesis angulares, 〈 〉, indican el promedio sobre el ensamble, lo que da La segunda igualdad requiere la equivalencia de partículas 1,…,N−1.La fórmula anterior es útil para relacionar g(r) con el factor de estructura estático S(q), definido como Los términos para los que i = j, suman en total una unidad, por lo que se puede escribir Usando las propiedades de la delta de Dirac tenemos de la ecuación anterior que y, por lo tanto, (5)lo que prueba la relación de Fourier mencionada anteriormente.Ya que esta información no es accesible experimentalmente, se puede sustraer de la ecuación y redefinir el factor de estructura como una función regular: Finalmente, podemos renombrar S(q) ≡ S′(q) y, si el sistema es un líquido, podemos invocar su isotropía: (6)Evaluando (6) en q = 0 y usando la relación entre la compresibilidad isotérmica χT y el factor de estructura en el origen, se obtiene la ecuación de compresibilidad: (7)La función de distribución radial es una medida importante ya que, a partir de ella se pueden calcular varias propiedades termodinámicas clave.Para sistemas tridimensionales, en donde las partículas interaccionan a través de potenciales por pares, la energía potencial del sistema puede calcularse de la siguiente manera:[3] donde N es el número de partículas del sistema, ρ es la densidad numérica y u(r) es el potencial a pares.Nótese que los resultados para la energía potencial y la presión no serán lo suficientemente precisos, comparados con cómo serían al calcularlos directamente, debido al promedio que se requiere en el cálculo de g(r).En sistemas diluidos (por ejemplo, los gases), las correlaciones de las posiciones de las partículas expresadas por g(r) se deben únicamente al potencial u(r) generado por la partícula de referencia, ignorando los efectos indirectos.En distancias r para las que u(r) es significativa, la densidad media local diferirá de la densidad media ρ, dependiendo del signo de u(r) (será mayor para interacciones negativas y menor para potenciales positivos).Esta similitud no es accidental; de hecho, al sustituir (12) en las relaciones anteriores de los parámetros termodinámicos —ecuaciones (7), (9) y (10)—, se obtienen las expansiones del virial correspondientes.[7] Nótese que ir desde la función S(q) experimental hasta g(r) no es directo y se requiere gran cantidad de análisis.[9] Esta técnica está limitada a partículas lo suficientemente grandes para ser detectadas ópticamente (en el rango de micras), pero tiene como ventaja que tiene resolución temporal.Asimismo, este tipo de obtención tiene resolución espacial (al nivel de partículas individuales), lo que permite revelar la morfología[11] y la dinámica de las estructuras locales en cristales coloidales,[12] vidrios,[13] geles,[14][15] y las interacciones hidrodinámicas.[16] Las funciones de correlación de orden superior, g(k), con k > 2, han sido menos estudiadas, puesto que generalmente son menos importantes para la termodinámica del sistema; además, no son accesibles con técnicas de dispersión convencionales.Sin embargo, estas funciones pueden ser medidas a través de dispersión de rayos X coherente y son interesantes puesto que revelan las simetrías locales en sistemas desordenados.
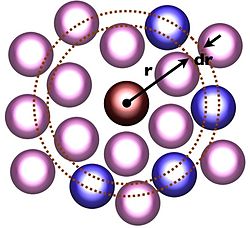
Cálculo de
g
(
r
). La partícula roja representa una partícula de referencia localizada en
O
, mientras que las partículas azules son aquella que se encuentran en posiciones
r
+ d
r
.