La púrpura trombocitopénica inmune ( PTI ), también conocida como púrpura trombocitopénica idiopática o trombocitopenia inmune , es un trastorno primario autoinmune de la hemostasia caracterizado por un recuento bajo de plaquetas en ausencia de otras causas. [1] [2] La PTI a menudo resulta en un mayor riesgo de sangrado de las superficies mucosas (como la nariz o las encías) o de la piel (lo que causa púrpura y hematomas ). [1] Dependiendo del grupo de edad afectado, la PTI causa dos síndromes clínicos distintos : una forma aguda que se observa en niños y una forma crónica en adultos. La PTI aguda a menudo sigue a una infección viral y generalmente es autolimitada (se resuelve en dos meses), mientras que la forma más crónica (que persiste durante más de seis meses) aún no tiene una causa específica identificada. Sin embargo, la patogénesis de la PTI es similar en ambos síndromes e involucra anticuerpos contra diversos antígenos de la superficie plaquetaria, como las glicoproteínas .
El diagnóstico de PTI implica identificar un recuento bajo de plaquetas mediante un hemograma completo , un análisis de sangre común . Sin embargo, dado que el diagnóstico se basa en excluir otras causas potenciales de un recuento bajo de plaquetas, en ciertos casos pueden ser necesarias investigaciones adicionales, como una biopsia de médula ósea .
Para casos leves, una observación cuidadosa puede ser suficiente. Sin embargo, en casos de recuentos de plaquetas muy bajos o sangrado significativo, las opciones de tratamiento pueden incluir corticosteroides , inmunoglobulina intravenosa , inmunoglobulina anti-D o medicamentos inmunosupresores . La PTI refractaria , que no responde al tratamiento convencional o muestra recaídas constantes después de la esplenectomía , requiere tratamiento para reducir el riesgo de hemorragia significativa. [3] Las transfusiones de plaquetas se pueden utilizar en casos graves con recuentos de plaquetas extremadamente bajos en personas que experimentan hemorragia. En algunos casos, el cuerpo puede compensar produciendo plaquetas anormalmente grandes .
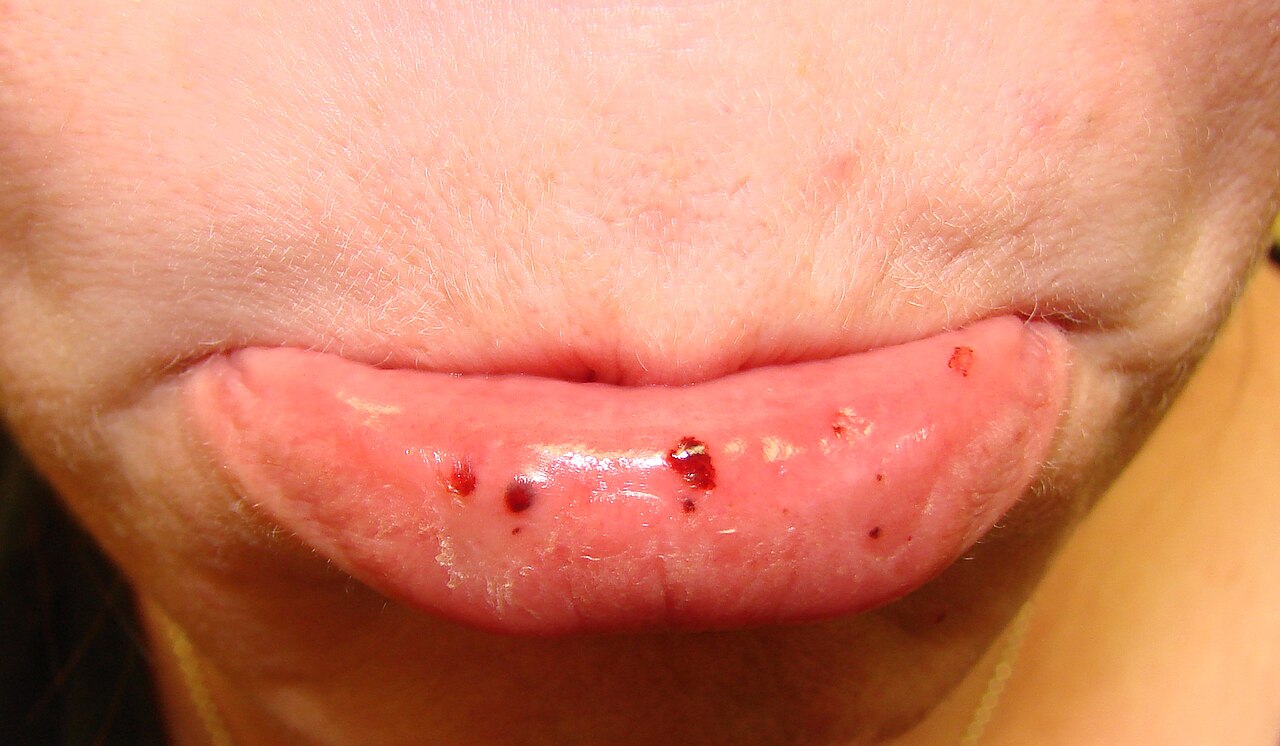
Los signos de PTI incluyen la formación espontánea de hematomas ( púrpura ) y petequias (pequeños hematomas), especialmente en las extremidades. Además, puede producirse sangrado de las fosas nasales y/o de las encías, así como menorragia (sangrado menstrual excesivo), si el recuento de plaquetas cae por debajo de 20.000 por μL. [4] Un recuento de plaquetas inferior a 10.000 por μL puede provocar la formación espontánea de hematomas (masas de sangre) en la boca o en otras membranas mucosas. Además, el tiempo de sangrado por laceraciones o abrasiones menores suele ser prolongado. [ cita necesaria ]
En los casos en que los recuentos de plaquetas caen a niveles extremadamente bajos (<5000 por μL), pueden surgir complicaciones graves y potencialmente mortales. Estas complicaciones incluyen hemorragia subaracnoidea o intracerebral (sangrado dentro del cráneo o el cerebro), hemorragia gastrointestinal baja u otras hemorragias internas. Una persona con PTI con un recuento de plaquetas extremadamente bajo es susceptible a sufrir una hemorragia interna como resultado de un traumatismo abdominal cerrado , como en un accidente automovilístico. Es poco probable que estas complicaciones ocurran cuando el recuento de plaquetas es inferior a 20 000 por μL. [5]
En aproximadamente el 60 por ciento de los casos se pueden detectar anticuerpos contra las plaquetas. [6] En la mayoría de los casos, estos anticuerpos son contra las glicoproteínas IIb-IIIa o Ib-IX de la membrana plaquetaria y son del tipo inmunoglobulina G (IgG). El experimento de Harrington-Hollingsworth estableció la patogénesis inmune de la PTI. [7]
El recubrimiento de las plaquetas con IgG las hace susceptibles a la opsonización y fagocitosis por parte de los macrófagos esplénicos , así como por las células de Kupffer en el hígado . También se cree que los autoanticuerpos IgG dañan los megacariocitos , las células precursoras de las plaquetas, aunque se cree que esto contribuye sólo ligeramente a la disminución del número de plaquetas. Investigaciones recientes ahora indican que la producción alterada de la hormona glicoproteica , la trombopoyetina , que es el estimulante de la producción de plaquetas, puede ser un factor que contribuya a la reducción de las plaquetas circulantes. Esta observación ha llevado al desarrollo de una clase de medicamentos dirigidos a la PTI denominados agonistas del receptor de trombopoyetina . [5]
El estímulo para la producción de autoanticuerpos en la PTI es probablemente una actividad anormal de las células T. [8] [9] [10] Los hallazgos preliminares sugieren que estas células T pueden verse influenciadas por medicamentos que se dirigen a las células B , como el rituximab . [11]

El diagnóstico de PTI es un proceso de exclusión. En primer lugar, se debe determinar que no hay anomalías sanguíneas aparte de un recuento bajo de plaquetas ni signos físicos aparte del sangrado. Luego, se deben excluir las causas secundarias (alrededor del 5 al 10 por ciento de los casos sospechosos de PTI), incluidos medicamentos ( quinina o heparina ), infección viral ( VIH o VHC ), enfermedades malignas ( leucemia ), enfermedades autoinmunes ( lupus eritematoso sistémico o síndrome antifosfolípido ), onyalai y otros. [4] [12] A todos los pacientes con presunta PTI se les deben realizar pruebas de detección del VIH y del virus de la hepatitis C, ya que los recuentos de plaquetas pueden corregirse mediante el tratamiento de la enfermedad subyacente. En aproximadamente del 2,7 al 5 por ciento de los casos coexisten anemia hemolítica autoinmune y PTI, una condición conocida como síndrome de Evans . [13] [14]
A pesar de la destrucción de las plaquetas por los macrófagos esplénicos, el bazo normalmente no está agrandado. De hecho, un bazo agrandado debería llevar a la búsqueda de otras posibles causas de la trombocitopenia. El tiempo de sangrado suele ser prolongado en pacientes con PTI. Sin embargo, las pautas prácticas de la Sociedad Estadounidense de Hematología [15] desaconsejan el uso del tiempo de sangrado en el diagnóstico y un tiempo de sangrado normal no excluye un trastorno plaquetario. [dieciséis]
El examen de médula ósea se puede realizar en pacientes mayores de 60 años y en aquellos que no responden al tratamiento, o cuando el diagnóstico es dudoso. [12] Al examinar la médula, se puede observar un aumento en la producción de megacariocitos y puede ayudar a establecer un diagnóstico de PTI. Un análisis de anticuerpos antiplaquetarios es una cuestión de preferencia del médico, ya que no hay acuerdo sobre si la especificidad del 80 por ciento de esta prueba es suficiente para ser clínicamente útil. [12]
Con raras excepciones, normalmente no es necesario realizar un tratamiento según el recuento de plaquetas. Muchas recomendaciones anteriores sugerían un cierto umbral de recuento de plaquetas (normalmente por debajo de 20,0/μL) como indicación de hospitalización o tratamiento. Las directrices actuales recomiendan el tratamiento sólo en casos de hemorragia importante. Las recomendaciones de tratamiento a veces difieren para la PTI adulta y pediátrica. [17]
El tratamiento inicial suele consistir en la administración de corticoides , un grupo de medicamentos que inhiben el sistema inmunológico. La dosis y el modo de administración están determinados por el recuento de plaquetas y si hay sangrado activo: en situaciones urgentes se pueden utilizar infusiones de dexametasona o metilprednisolona , mientras que en casos menos graves pueden ser suficientes prednisona o prednisolona oral. Una vez que el recuento de plaquetas ha mejorado, la dosis de esteroide se reduce gradualmente mientras se controla la posibilidad de recaída. Entre el 60 y el 90 por ciento experimentará una recaída durante la reducción o el cese de la dosis. [12] [18] Si es posible, se evitan los esteroides a largo plazo debido a posibles efectos secundarios que incluyen osteoporosis , diabetes y cataratas . [19]
Otra opción, adecuada para pacientes Rh positivos con bazos funcionales, es la administración intravenosa de inmunoglobulina Rho(D) [humana; Anti-D]. El mecanismo de acción del anti-D no se comprende completamente. Sin embargo, después de la administración, los complejos de glóbulos rojos recubiertos con anti-D saturan los sitios del receptor Fcγ en los macrófagos , lo que produce una destrucción preferencial de los glóbulos rojos (RBC), preservando así las plaquetas recubiertas de anticuerpos . Hay dos productos anti-D indicados para su uso en pacientes con PTI: WinRho SDF y Rhophylac. Las reacciones adversas más comunes son dolor de cabeza (15%), náuseas/vómitos (12%), escalofríos (<2%) y fiebre (1%). [ cita necesaria ]
Cada vez se utilizan más inmunosupresores como el micofenolato de mofetilo y la azatioprina debido a su eficacia. En casos refractarios crónicos, donde se ha confirmado la patogénesis inmune, [20] se puede intentar el uso no autorizado del alcaloide de la vinca [21] [22] [23] y el agente quimioterapéutico vincristina . [24] [25] Sin embargo, la vincristina tiene efectos secundarios importantes [26] y su uso en el tratamiento de la PTI debe abordarse con precaución, especialmente en niños.
En algunos casos, se puede infundir inmunoglobulina intravenosa (IgIV) para disminuir la velocidad a la que los macrófagos consumen plaquetas marcadas con anticuerpos . Sin embargo, aunque a veces es eficaz, es costoso y produce una mejora que generalmente dura menos de un mes. Sin embargo, en el caso de un paciente con PTI ya programado para cirugía que tiene un recuento de plaquetas peligrosamente bajo y ha experimentado una mala respuesta a otros tratamientos, la IgIV puede aumentar rápidamente el recuento de plaquetas y también puede ayudar a reducir el riesgo de hemorragia grave al aumentar transitoriamente recuentos de plaquetas. [27]
Los agonistas del receptor de trombopoyetina son agentes farmacéuticos que estimulan la producción de plaquetas en la médula ósea. En esto se diferencian de los agentes comentados anteriormente que actúan intentando reducir la destrucción plaquetaria. [28] Actualmente se encuentran disponibles dos de estos productos:
Los agonistas del receptor de trombopoyetina han mostrado el mayor éxito hasta el momento en el tratamiento de pacientes con PTI refractaria. [34]
Los efectos secundarios de los agonistas del receptor de trombopoyetina incluyen dolor de cabeza, dolor articular o muscular, mareos, náuseas o vómitos y un mayor riesgo de coágulos sanguíneos. [28]
Se puede considerar la esplenectomía (extirpación del bazo ) en pacientes que no responden al tratamiento con esteroides, tienen recaídas frecuentes o no pueden reducirse gradualmente los esteroides después de unos meses. Las plaquetas que han sido unidas por anticuerpos son absorbidas por los macrófagos del bazo (que tienen receptores Fc ), por lo que la eliminación del bazo reduce la destrucción de las plaquetas. El procedimiento es potencialmente riesgoso en casos de PTI debido a la mayor posibilidad de sangrado significativo durante la cirugía. La remisión duradera después de la esplenectomía se logra en entre el 60 y el 80 por ciento de los casos de PTI. [35] Aunque existe un consenso sobre la eficacia a corto plazo de la esplenectomía, los hallazgos sobre su eficacia a largo plazo y sus efectos secundarios son controvertidos. [34] [36] Después de la esplenectomía, entre el 11,6 y el 75 por ciento de los casos de PTI recayeron y entre el 8,7 y el 40 por ciento de los casos de PTI no tuvieron respuesta a la esplenectomía. [34] [37] [38] [39] El uso de la esplenectomía para tratar la PTI ha disminuido desde el desarrollo de la terapia con esteroides y otros remedios farmacéuticos. [40]
Normalmente no se recomienda la transfusión de plaquetas sola, excepto en casos de emergencia, y por lo general no logra producir un aumento del recuento de plaquetas a largo plazo. Esto se debe a que el mecanismo autoinmune subyacente que destruye las plaquetas del paciente también destruirá las plaquetas del donante, por lo que las transfusiones de plaquetas no se consideran una opción de tratamiento a largo plazo. [41]
En adultos, particularmente aquellos que viven en áreas con una alta prevalencia de Helicobacter pylori (que normalmente habita en la pared del estómago y se ha asociado con úlceras pépticas ), se ha demostrado que la identificación y el tratamiento de esta infección mejoran los recuentos de plaquetas en un tercio de los pacientes. En un quinto, el recuento de plaquetas se normalizó por completo; esta tasa de respuesta es similar a la encontrada en el tratamiento con rituximab, que es más caro y menos seguro. [42] En los niños, este enfoque no está respaldado por evidencia, excepto en áreas de alta prevalencia. Las pruebas de urea en el aliento y las pruebas de antígenos en heces funcionan mejor que las pruebas serológicas ; además, la serología puede dar resultados falsos positivos después del tratamiento con IVIG. [43]
En general, los pacientes con PTI aguda rara vez tendrán hemorragias que pongan en peligro su vida. [48] la mayoría de los pacientes finalmente tienen recuentos de plaquetas estables pero más bajos, lo cual es hemostático para una persona. A diferencia de los pacientes pediátricos que pueden curarse, la mayoría de los adultos seguirán un curso crónico incluso después de la esplenectomía. [49]
Se considera que un recuento normal de plaquetas está en el rango de 150 000 a 450 000 por microlitro (μL) de sangre para la mayoría de las personas sanas. Por lo tanto, uno puede considerarse trombocitopénico por debajo de ese rango, aunque el umbral para un diagnóstico de PTI no está vinculado a ningún número específico. [50]
La incidencia de la PTI se estima en 50 a 100 casos nuevos por millón por año, y la mitad de esa cifra corresponde a los niños. Al menos el 70 por ciento de los casos infantiles terminarán en remisión dentro de seis meses, incluso sin tratamiento. [51] [52] [53] Además, un tercio de los casos crónicos restantes generalmente remiten durante la observación de seguimiento, y otro tercio terminará con solo trombocitopenia leve (definida como un recuento de plaquetas superior a 50 000). [51] Se ha identificado una serie de genes y polimorfismos relacionados con el sistema inmunitario que influyen en la predisposición a la PTI; el alelo FCGR3a-V158 y KIRDS2/DL2 aumentan la susceptibilidad y se ha demostrado que KIR2DS5 es protector. [54] [55]
La PTI suele ser crónica en adultos [56] y la probabilidad de una remisión duradera es del 20 al 40 por ciento. [18] La proporción entre hombres y mujeres en el grupo de adultos varía de 1:1,2 a 1,7 en la mayoría de los rangos de edad (los casos infantiles son aproximadamente iguales para ambos sexos) y la edad promedio de los adultos en el momento del diagnóstico es de 56 a 60 años. [12] La proporción entre casos de hombres y mujeres en adultos tiende a ampliarse con la edad. En los Estados Unidos, se cree que la población adulta crónica es de aproximadamente 60 000 personas, y las mujeres superan en número a los hombres aproximadamente 2 a 1, lo que ha dado lugar a que la PTI sea designada enfermedad huérfana . [57]
La tasa de mortalidad debida a la PTI crónica varía, pero tiende a ser más alta en relación con la población general para cualquier rango de edad. En un estudio realizado en Gran Bretaña , se observó que la PTI causa una tasa de mortalidad aproximadamente un 60 por ciento más alta en comparación con sujetos del mismo sexo y edad sin PTI. Este mayor riesgo de muerte con PTI se concentra en gran medida en personas de mediana edad y ancianos . El noventa y seis por ciento de las muertes relacionadas con la PTI reportadas fueron personas de 45 años o más. No se observaron diferencias significativas en la tasa de supervivencia entre hombres y mujeres. [58]
La incidencia de la PTI en el embarazo no se conoce bien. Puede ocurrir durante cualquier trimestre del embarazo. Es la causa más común de trombocitopenia significativa (menos de 100.000 plaquetas) en el segundo trimestre, y es una causa común de trombocitopenia significativa en el primer y tercer trimestre. [59] Al igual que en personas no embarazadas, la PTI en el embarazo es un diagnóstico de exclusión y otras posibles causas de niveles bajos de plaquetas en el embarazo requieren consideración. Estas incluyen causas obstétricas como preeclampsia , síndrome HELLP (hemólisis, enzimas hepáticas elevadas y plaquetas bajas) o microangiopatías trombóticas que pueden ocurrir durante el embarazo. [59] También se deben descartar otras causas de trombocitopenia que pueden ocurrir durante el embarazo, como la trombocitopenia inducida por fármacos , la trombocitopenia hereditaria y la pseudotrombocitopenia. [59] La PTI puede ser difícil de distinguir de la trombocitopenia gestacional (que es, con mucho, la causa más común de trombocitopenia en el embarazo). A diferencia de la PTI, el recuento de plaquetas en la trombocitopenia gestacional rara vez desciende por debajo de 100.000, y un recuento de plaquetas por debajo de 80.000 es aún más raro (observado en menos del 0,1% de los casos de trombocitopenia gestacional). Además, a diferencia de la PTI, la trombocitopenia gestacional no es una causa de hemorragia materna o neonatal, ni de trombocitopenia neonatal. [59]
Las mujeres con PTI suelen tener una disminución en el recuento de plaquetas cuando quedan embarazadas, lo que a menudo requiere tratamiento. [59] Las mujeres embarazadas con PTI tienen 1,83 veces más probabilidades de tener episodios de sangrado durante el embarazo en comparación con las mujeres no embarazadas con PTI; sin embargo, con el tratamiento adecuado, las plaquetas rara vez caen por debajo de 30.000. [59] En la PTI, el sangrado severo es una ocurrencia rara y, con tratamiento, las muertes maternas debido a la PTI son extremadamente raras. [59] No se ha encontrado que la PTI aumente el riesgo de algunas complicaciones obstétricas comunes; sin que se haya observado un mayor riesgo de preeclampsia, parto prematuro, desprendimiento de placenta o coágulos de sangre . Además, en aquellas personas con PTI, los recuentos de plaquetas suelen volver a los niveles previos al embarazo después del parto. [59]
Los autoanticuerpos antiplaquetarios en una mujer embarazada con PTI atacarán las plaquetas del propio paciente y también cruzarán la placenta y reaccionarán contra las plaquetas fetales, lo que provocará trombocitopenia. Los niveles de trombopoyetina también aumentan durante el embarazo (lo que se cree que se debe a la producción placentaria de estradiol ) y este estradiol conduce a una disminución de la actividad de los megacariocitos y, por lo tanto, a una disminución de la producción de plaquetas. [59] Por lo tanto, la PTI es una causa importante de trombocitopenia inmunitaria fetal y neonatal. Aproximadamente el 10% de los recién nacidos afectados por PTI tendrán recuentos de plaquetas <50 000/uL y entre el 1% y el 2% tendrán un riesgo de hemorragia intracerebral, comparable al de los lactantes con trombocitopenia aloinmune neonatal (NAIT). [60] [61]
Ninguna prueba de laboratorio puede predecir de manera confiable si ocurrirá trombocitopenia neonatal. El riesgo de trombocitopenia neonatal aumenta con: [62]
Se recomienda que las mujeres embarazadas con trombocitopenia o con un diagnóstico previo de PTI se realicen pruebas de anticuerpos antiplaquetarios séricos. Las indicaciones para el tratamiento de personas embarazadas con PTI incluyen la presencia de sangrado, recuentos de plaquetas inferiores a 20 000-30 000, procedimientos planificados (como una amniocentesis ) y elevación de los niveles de plaquetas antes del parto (el nivel mínimo de plaquetas para un parto vaginal es 30 000 y por cesárea es 50.000). [59] En raras ocasiones, puede ser necesaria una transfusión de plaquetas para facilitar el parto seguro. [59] Las terapias de primera línea para la PTI incluyen esteroides como prednisona , IVIG o ambos. Las terapias de segunda línea (generalmente utilizadas en aquellos en quienes las terapias de primera línea fallan y persiste la trombocitopenia) incluyen rituximab , agonistas del receptor de trombopoetina (como romiplostim ) o una esplenectomía . [59] Se pueden realizar transfusiones de plaquetas en recién nacidos, según el grado de trombocitopenia. También se pueden administrar IVIG y esteroides ( metilprednisolona ) para aumentar el recuento de plaquetas. [59] Se recomienda que los recién nacidos sean seguidos con recuentos seriados de plaquetas durante los primeros días después del nacimiento. [60] [62] También se recomiendan ecografías o resonancias magnéticas en bebés con trombocitopenia grave para descartar una hemorragia cerebral . [59]
Después de los informes iniciales del médico portugués Amato Lusitano en 1556 y de Lázaro de la Rivière (médico del rey de Francia) en 1658, fue el médico y poeta alemán Paul Gottlieb Werlhof quien en 1735 escribió el informe inicial más completo sobre la púrpura de PTI. En aquel momento se desconocían las plaquetas. [63] El nombre "enfermedad de Werlhof" se utilizó más ampliamente antes de que el nombre descriptivo actual se hiciera más popular. [63] [64] Las plaquetas se describieron a principios del siglo XIX y en la década de 1880 varios investigadores relacionaron la púrpura con anomalías en el recuento de plaquetas. [63] [65] El primer informe de una terapia exitosa para la PTI fue en 1916, cuando un joven estudiante de medicina polaco , Paul Kaznelson , describió la respuesta de una paciente a una esplenectomía . [63] La esplenectomía siguió siendo un remedio de primera línea hasta la introducción de la terapia con esteroides en la década de 1950. [63]


