Las talasemias beta ( β talasemias ) son un grupo de trastornos hereditarios de la sangre . Son formas de talasemia causadas por la síntesis reducida o ausente de las cadenas beta de la hemoglobina que resultan en resultados variables que van desde anemia grave hasta individuos clínicamente asintomáticos. La incidencia anual global se estima en uno en 100.000. [4] Las talasemias beta ocurren debido a disfunciones en la subunidad beta de la hemoglobina o HBB. La gravedad de la enfermedad depende de la naturaleza de la mutación. [5]
La incapacidad del cuerpo para construir nuevas cadenas beta conduce a una producción insuficiente de HbA (hemoglobina adulta). [6] El desequilibrio de las cadenas de globina alfa y beta conduce a una eritropoyesis ineficaz , un aumento de la hemólisis y una alteración de la homeostasis del hierro . [7] Los pacientes pueden requerir transfusiones de sangre repetidas a lo largo de la vida para mantener niveles suficientes de hemoglobina. En consecuencia, los pacientes también pueden desarrollar problemas graves asociados con la sobrecarga de hierro . [4]
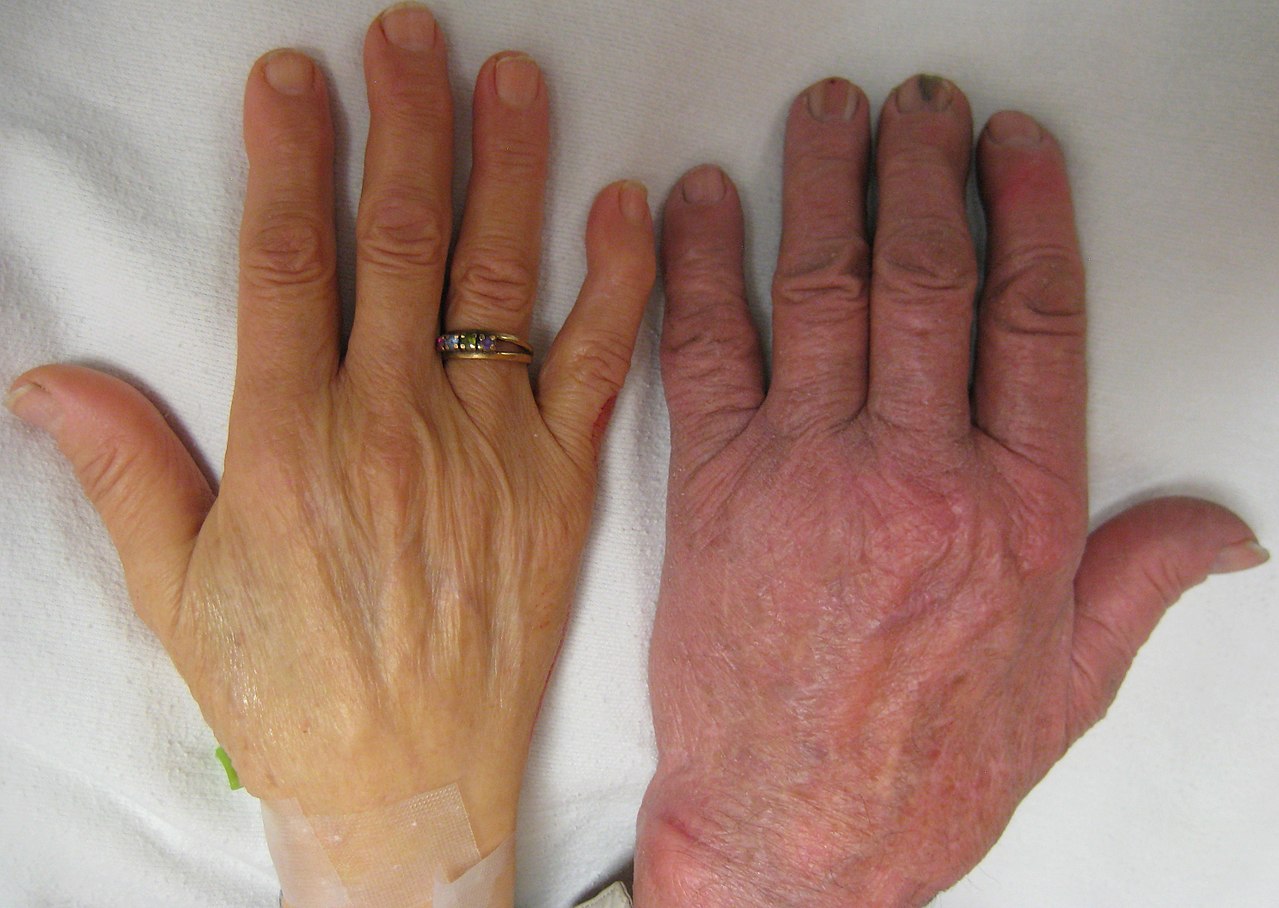
Se han descrito tres formas principales: talasemia menor, talasemia intermedia y talasemia mayor, que varían desde asintomática o con síntomas leves hasta anemia grave que requiere transfusiones de por vida. [8] Los individuos con beta talasemia mayor (aquellos que son homocigotos para mutaciones de talasemia o que heredan 2 mutaciones) generalmente presentan dentro de los primeros dos años de vida anemia grave sintomática, crecimiento deficiente y anomalías esqueléticas. La talasemia mayor no tratada eventualmente conduce a la muerte, generalmente por insuficiencia cardíaca ; por lo tanto, el cribado prenatal es muy importante. [9] Aquellos con beta talasemia intermedia (aquellos que son heterocigotos compuestos para la mutación de beta talasemia) generalmente presentan más tarde en la vida síntomas de anemia leves a moderados. [8] El rasgo de beta talasemia (también conocido como beta talasemia menor) implica herencia heterocigótica de una mutación de beta talasemia y los pacientes generalmente tienen microcitosis con anemia hipocrómica limítrofe y generalmente son asintomáticos o tienen síntomas leves. [8] La beta talasemia menor también puede presentarse como portadores silenciosos de beta talasemia; aquellos que heredan una mutación beta talasémica pero no presentan anomalías hematológicas ni síntomas. [8] Algunas personas con talasemia son susceptibles a complicaciones de salud que involucran el bazo (hiperesplenismo) y cálculos biliares (debido a hiperbilirrubinemia por hemólisis periférica). [8] [1] Estas complicaciones se encuentran principalmente en pacientes con talasemia mayor e intermedia. [ cita requerida ]
El exceso de hierro (por hemólisis o transfusiones ) causa complicaciones graves en el hígado, el corazón y las glándulas endocrinas . Los síntomas graves incluyen cirrosis hepática , fibrosis hepática y, en casos extremos, cáncer de hígado . [10] La insuficiencia cardíaca, el retraso del crecimiento, la diabetes y la osteoporosis son afecciones potencialmente mortales que pueden ser causadas por la beta talasemia mayor. [11] Las principales anomalías cardíacas observadas como resultado de la beta talasemia y la sobrecarga de hierro incluyen disfunción sistólica y diastólica del ventrículo izquierdo, hipertensión pulmonar, valvulopatía, arritmias y pericarditis. Se observa un aumento de la absorción gastrointestinal de hierro en todos los grados de beta talasemia, y una mayor destrucción de glóbulos rojos por parte del bazo debido a una eritropoyesis ineficaz libera aún más hierro en el torrente sanguíneo . [12]
Los síntomas adicionales de la talasemia beta mayor o intermedia incluyen los síntomas clásicos de anemia moderada a severa, incluyendo fatiga, retraso del crecimiento y desarrollo en la niñez, úlceras en las piernas y falla orgánica. [8] La eritropoyesis ineficaz (producción de glóbulos rojos) también puede conducir a una expansión compensatoria de la médula ósea que luego puede conducir a cambios/deformidades óseas, dolor óseo y anomalías craneofaciales. [8] Los órganos extramedulares como el hígado y el bazo que también pueden sufrir eritropoyesis se activan dando lugar a hepatoesplenomegalia (agrandamiento del hígado y el bazo). [8] Otros tejidos en el cuerpo también pueden convertirse en sitios de eritropoyesis, dando lugar a pseudotumores hematopoyéticos extramedulares que pueden causar síntomas compresivos si ocurren en la cavidad torácica o el canal espinal. [8]
Se pueden distinguir dos grupos principales de mutaciones:
Las mutaciones se caracterizan como (βo) si impiden cualquier formación de cadenas de globina β, las mutaciones se caracterizan como (β+) si permiten que se produzca alguna formación de cadenas de globina β. [8]
Para las decisiones de tratamiento y la elegibilidad para ensayos clínicos, se utilizan las categorías de talasemia dependiente de transfusión (TDT) y talasemia no dependiente de transfusión (NTDT). Por lo general, se considera que los pacientes tienen NTDT si han recibido menos de 6 unidades de glóbulos rojos en los últimos 6 meses y ninguna en los 2 meses anteriores. [18]

La beta talasemia es una enfermedad hereditaria que afecta a la hemoglobina. Como ocurre con aproximadamente la mitad de todas las enfermedades hereditarias, [19] una mutación hereditaria daña el ensamblaje del ARN mensajero (ARNm) que se transcribe a partir de un cromosoma . El ADN contiene tanto las instrucciones ( genes ) para unir aminoácidos en proteínas , como también tramos de ADN que desempeñan papeles importantes en la regulación de los niveles de proteína producida . [20]
En la talasemia, se incluye en el ARNm una longitud contigua adicional o un fragmento discontinuo de instrucciones no codificantes. Esto sucede porque la mutación borra el límite entre las porciones intrónica y exónica de la plantilla de ADN. [21] Debido a que todas las secciones codificantes pueden seguir presentes, se puede producir hemoglobina normal y el material genético añadido, si produce patología, en cambio altera las funciones reguladoras lo suficiente como para producir anemia. Las subunidades alfa y beta normales de la hemoglobina tienen cada una una porción central que contiene hierro (hemo) que permite que la cadena proteica de una subunidad se pliegue a su alrededor. La hemoglobina adulta normal contiene 2 subunidades alfa y 2 beta. [22] Las talasemias generalmente afectan solo a los ARNm para la producción de las cadenas beta (de ahí el nombre). Dado que la mutación puede ser un cambio en solo una base ( polimorfismo de un solo nucleótido ), los esfuerzos en curso buscan terapias genéticas para hacer esa única corrección. [23] [24]
Los antecedentes familiares y la ascendencia son factores que aumentan el riesgo de padecer beta talasemia. Según los antecedentes familiares, si los padres o abuelos de una persona tuvieron beta talasemia mayor o intermedia, existe una probabilidad del 75 % (3 de 4) (consulte el cuadro de herencia en la parte superior de la página) de que un hijo herede el gen mutado. Incluso si un hijo no tiene beta talasemia mayor o intermedia, puede ser portador, lo que posiblemente dé como resultado que las generaciones futuras de su descendencia tengan beta talasemia. [ cita requerida ]
Otro factor de riesgo es la ascendencia. La beta talasemia se presenta con mayor frecuencia en personas de ascendencia italiana, griega, de Oriente Medio, del sur de Asia y africana. [25]

El dolor abdominal debido a hiperesplenismo , infarto esplénico y dolor en el cuadrante superior derecho causado por cálculos biliares son manifestaciones clínicas importantes. Sin embargo, diagnosticar la talasemia solo a partir de los síntomas es inadecuado. Los médicos notan estos signos como asociativos debido a la complejidad de esta enfermedad. [26] Los siguientes signos asociativos pueden dar fe de la gravedad del fenotipo : palidez, crecimiento deficiente, ingesta inadecuada de alimentos, esplenomegalia , ictericia , hiperplasia maxilar, maloclusión dental , colelitiasis , soplo de eyección sistólico en presencia de anemia grave y fracturas patológicas. Según los síntomas, se solicitan pruebas para un diagnóstico diferencial . Estas pruebas incluyen hemograma completo ; electroforesis de hemoglobina ; transferrina sérica , ferritina , capacidad total de unión al hierro ; urobilina y urobilógeno en orina; frotis de sangre periférica , que puede mostrar codocitos o células diana; [27] hematocrito ; y bilirrubina sérica. [28] [29] El patrón esperado en la electroforesis de hemoglobina en personas con beta-talasemia es un nivel aumentado de hemoglobina A2 y un nivel ligeramente aumentado de hemoglobina F. [ cita requerida ] El diagnóstico se confirma con electroforesis de hemoglobina o cromatografía líquida de alto rendimiento. [8]
Cambios esqueléticos asociados con la expansión de la médula ósea:
Todas las beta talasemias pueden presentar glóbulos rojos anormales; se realiza un seguimiento de los antecedentes familiares mediante un análisis de ADN. [3] Esta prueba se utiliza para investigar deleciones y mutaciones en los genes productores de alfa y beta globina. Se pueden realizar estudios familiares para evaluar el estado de portador y los tipos de mutaciones presentes en otros miembros de la familia. Las pruebas de ADN no son rutinarias, pero pueden ayudar a diagnosticar la talasemia y determinar el estado de portador. En la mayoría de los casos, el médico tratante utiliza un prediagnóstico clínico que evalúa los síntomas de anemia: fatiga, disnea y poca tolerancia al ejercicio. [30] El análisis genético adicional puede incluir HPLC si la electroforesis de rutina resulta difícil. [28]
La beta talasemia es una enfermedad hereditaria que permite un tratamiento preventivo mediante el cribado de portadores y el diagnóstico prenatal. Se puede prevenir si uno de los padres tiene genes normales, lo que da lugar a pruebas de detección que permiten a los portadores seleccionar parejas con hemoglobina normal. Un estudio tuvo como objetivo detectar los genes que podrían dar lugar a una descendencia con enfermedad de células falciformes. Los pacientes diagnosticados con beta talasemia tienen MCH ≤ 26 pg y un RDW < 19. De 10.148 pacientes, 1.739 pacientes tenían un fenotipo de hemoglobina y RDW compatible con beta talasemia. Después de la selección de pacientes, se analizaron los niveles de HbA2 y se presentaron 77 pacientes con beta talasemia. [31] Este procedimiento de cribado resultó insensible en poblaciones de ascendencia de África occidental debido a que los indicadores tienen una alta prevalencia de alfa talasemia. Los países tienen programas que distribuyen información sobre los riesgos reproductivos asociados a los portadores de hemoglobinopatías. Los programas de detección de portadores de talasemia incluyen programas educativos en escuelas, fuerzas armadas y a través de los medios de comunicación, así como también asesoramiento a los portadores y parejas de portadores. [32] La detección ha demostrado una incidencia reducida; en 1995, la prevalencia en Italia se redujo de 1:250 a 1:4000, y una disminución del 95% en esa región. La disminución de la incidencia ha beneficiado a los afectados por talasemia, ya que la demanda de sangre ha disminuido, mejorando así el suministro de tratamiento. [ cita requerida ]

Los niños afectados requieren transfusiones de sangre regulares durante toda su vida . Los trasplantes de médula ósea pueden ser curativos para algunos niños. [33] Los pacientes reciben transfusiones de sangre frecuentes que provocan o potencian la sobrecarga de hierro . [34] El tratamiento de quelación del hierro es necesario para prevenir daños a los órganos internos en casos de sobrecarga de hierro. Los avances en los tratamientos de quelación del hierro permiten a los pacientes con talasemia mayor vivir una vida larga con acceso al tratamiento adecuado. Los quelantes populares incluyen la deferoxamina y la deferiprona . [35] [36]
El quelante oral deferasirox fue aprobado para su uso en 2005 en algunos países. [37] [38] El trasplante de médula ósea es la única cura y está indicado para pacientes con talasemia mayor grave. El trasplante puede eliminar la dependencia de un paciente de las transfusiones. A falta de un donante compatible, se puede concebir un hermano salvador mediante diagnóstico genético preimplantacional (DGP) que esté libre de la enfermedad y que sea compatible con el tipo de antígeno leucocitario humano (HLA) del receptor. [39]
La ferritina sérica (la forma de almacenamiento del hierro) se mide de forma rutinaria en pacientes con beta talasemia para determinar el grado de sobrecarga de hierro; los niveles elevados de ferritina orientan el uso de terapia de quelación del hierro. Los tres quelantes de hierro, deferoxamina subcutánea, deferiprona oral y deferasirox oral, se pueden utilizar como monoterapia o en combinación; se ha demostrado que todos ellos reducen los niveles de hierro sérico/sistémico, hepático y cardíaco, así como el riesgo de arritmia cardíaca, insuficiencia cardíaca y muerte. [8] La resonancia magnética hepática y miocárdica también se utiliza para cuantificar la deposición de hierro en los órganos diana, especialmente el corazón y el hígado, para guiar la terapia. [8]
Los científicos del Weill Cornell Medical College han desarrollado una estrategia de terapia génica que podría tratar tanto la beta-talasemia como la anemia falciforme. La tecnología se basa en la administración de un vector lentiviral que lleva tanto el gen de la β-globina humana como un aislador de anquirina para mejorar la transcripción y traducción de genes y aumentar los niveles de producción de β-globina. [40]
El 10 de junio de 2022, un panel asesor federal de EE. UU. recomendó que la FDA aprobara un tratamiento de terapia génica para su uso en la beta talasemia. [41] El fabricante Bluebird bio cobra 2,8 millones de dólares en Estados Unidos por su tratamiento único Zynteglo ( betibeglogene autotemcel ). [42] [43]
Se han desarrollado terapias de edición genética destinadas a aumentar la producción de hemoglobina fetal en la beta talasemia, así como en la anemia de células falciformes, inhibiendo el gen BCL11A . [44] [45] Exagamglogene autotemcel , vendido bajo la marca Casgevy, es una terapia genética para el tratamiento de la beta talasemia dependiente de transfusiones desarrollada por Vertex Pharmaceuticals y CRISPR Therapeutics . [46]
El tratamiento fue aprobado en el Reino Unido para el tratamiento de la beta talasemia dependiente de transfusión en noviembre de 2023 [47] [48] [49] y en los Estados Unidos en enero de 2024. [50] [51] [52]
Exagamglogene autotemcel es el primer tratamiento de terapia génica basado en células que utiliza la tecnología de edición genética CRISPR/Cas9 aprobado por la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA). [50]
La terapia génica se realiza a partir de las propias células madre de la sangre del receptor, que se modifican y se administran en una infusión única de una sola dosis como parte de un trasplante de células madre hematopoyéticas . Antes del tratamiento, se recolectan las propias células madre del receptor y luego el receptor debe someterse a un acondicionamiento mieloablativo (quimioterapia de dosis alta), un proceso que extrae células de la médula ósea para que puedan reemplazarse con las células modificadas en exagamglogene autotemcel . [ cita requerida ]
Los pacientes con talasemia mayor son más propensos a someterse a una esplenectomía. El uso de esplenectomías ha disminuido en los últimos años debido a la menor prevalencia de hiperesplenismo en pacientes adecuadamente transfundidos. La esplenectomía también se asocia con un mayor riesgo de infecciones y una mayor morbilidad debido a la enfermedad vascular, ya que el bazo participa en la limpieza para eliminar del cuerpo los glóbulos rojos patológicos o anormales. [8] Los pacientes con hiperesplenismo tienen más probabilidades de tener una menor cantidad de células sanguíneas sanas en su cuerpo de lo normal y presentar síntomas de anemia. Las diferentes técnicas quirúrgicas son el método abierto y el laparoscópico. [2] El método laparoscópico requiere un tiempo de operación más largo, pero un período de recuperación más corto con una cicatriz quirúrgica más pequeña y menos prominente. Si no es necesario extirpar todo el bazo, puede realizarse una esplenectomía parcial; este método preserva parte de la función inmunológica al tiempo que reduce la probabilidad de hiperesplenismo. Aquellos que se sometan a una esplenectomía deben recibir una vacuna antineumocócica adecuada al menos una semana (preferiblemente tres semanas) antes de la cirugía. [53]
La terapia transfusional a largo plazo (en pacientes con beta talasemia dependiente de transfusión) es un tratamiento utilizado para mantener los niveles de hemoglobina en un nivel de hemoglobina pretransfusional objetivo de 9-10,5 g/dl (11-12 g/dl en pacientes con cardiopatía concomitante). [8] Para garantizar transfusiones de sangre de calidad, los glóbulos rojos concentrados deben ser leucorreducidos. Al tener paquetes de sangre leucorreducidos, el paciente tiene un menor riesgo de desarrollar reacciones adversas por glóbulos blancos contaminados y previene la aloinmunización plaquetaria. [54] Los pacientes con reacciones alérgicas a las transfusiones o anticuerpos inusuales contra los glóbulos rojos deben recibir glóbulos rojos lavados o glóbulos rojos criopreservados. Los glóbulos rojos lavados han sido eliminados de las proteínas plasmáticas que se habrían convertido en un objetivo de los anticuerpos del paciente, lo que permite que la transfusión se realice de manera segura. Los glóbulos rojos criopreservados se utilizan para mantener un suministro de unidades de donantes raros para pacientes con anticuerpos inusuales contra los glóbulos rojos o falta de antígenos comunes de glóbulos rojos. Estas transfusiones regulares promueven el crecimiento normal y las actividades físicas, y suprimen la hiperactividad de la médula ósea y la hematopoyesis extramedular, ayudando a reducir las masas dolorosas en algunos casos. [18] Sin embargo, estos beneficios deben sopesarse frente a las consideraciones de sobrecarga de hierro, y se deben realizar ajustes a medida que avanza el curso clínico. [18]
Durante la homeostasis normal del hierro, el hierro circulante se une a la transferrina. Pero con la sobrecarga de hierro (como en las transfusiones de sangre frecuentes), la capacidad de la transferrina para unirse al hierro se excede y el hierro no unido a la transferrina se acumula. Este hierro no unido es tóxico debido a su alta propensión a inducir especies de oxígeno y es responsable del daño celular. La prevención de la sobrecarga de hierro protege a los pacientes de la morbilidad y la mortalidad. El objetivo principal es unirse al hierro y eliminarlo del cuerpo a una tasa igual a la tasa de entrada de hierro transfusional o mayor que la entrada de hierro. [55] La quelación del hierro es una terapia médica que puede prevenir las complicaciones de la sobrecarga de hierro. [8] Cada unidad de sangre transfundida contiene 200-250 mg de hierro y el cuerpo no tiene un mecanismo natural para eliminar el exceso de hierro. El exceso de hierro puede eliminarse con quelantes de hierro (deferoxamina, deferiprona y deferasirox). [56] La concentración de hierro en el hígado (LIC) puede medirse mediante imágenes por resonancia magnética (IRM) R2 o T2*. En las partes del mundo donde no se dispone de técnicas avanzadas de resonancia magnética, se puede utilizar la ferritina sérica como alternativa. En pacientes con sobrecarga de hierro, es razonable iniciar la terapia de quelación si la LIC es >5 mg Fe por gramo de peso seco (ps) o el nivel de ferritina sérica es >800 ng/ml. La terapia de quelación puede interrumpirse cuando la LIC es <3 mg Fe por gramo de ps o la ferritina sérica <300 ng/ml. [18]
Luspatercept (ACE-536) es una proteína de fusión recombinante que se utiliza como tratamiento en adultos con beta talasemia dependiente de transfusión. Consiste en un dominio extracelular modificado del receptor de activina humano tipo IIB unido a la porción Fc del anticuerpo IgG1 humano. [8] La molécula se une a determinados ligandos de la superfamilia del factor de crecimiento transformante beta para bloquear la señalización SMAD2 y 3 , mejorando así la maduración eritroide. [8] Se ha demostrado que el medicamento reduce la carga transfusional en un 33 % en adultos con beta talasemia dependiente de transfusión en comparación con placebo y también se asoció con una disminución de los niveles de ferritina (sin disminuciones significativas de los niveles de hierro hepático o cardíaco). [8]
Los pacientes con beta talasemia intermedia suelen presentar anemia leve a moderada (7-10 g/dl) a los 2 años de edad. [18] Es posible que no requieran transfusiones o que requieran transfusiones de sangre episódicas en determinadas circunstancias (infección, embarazo, cirugía). [8] Los pacientes con transfusiones frecuentes pueden desarrollar sobrecarga de hierro y requerir terapia de quelación . [57] La transmisión es autosómica recesiva ; sin embargo, se han descrito mutaciones dominantes y heterocigotos compuestos . Se recomienda asesoramiento genético y se puede ofrecer diagnóstico prenatal . [58]
Los pacientes con beta talasemia menor suelen ser asintomáticos y a menudo se los controla sin tratamiento. [8] La beta talasemia menor puede coexistir con otras afecciones como la hepatitis B crónica , la hepatitis C crónica , la enfermedad del hígado graso no alcohólico y la enfermedad hepática alcohólica que, cuando se combinan o coexisten, pueden hacer que una persona tenga una sobrecarga de hierro en el hígado y una enfermedad hepática más grave. [59]
La forma beta de la talasemia es particularmente frecuente entre los pueblos mediterráneos y esta asociación geográfica es responsable de su denominación: thalassa ( θάλασσα ) es la palabra griega para mar y haima ( αἷμα ) es la palabra griega para sangre. [60] [61] [ cita requerida ] En Europa , las concentraciones más altas de la enfermedad se encuentran en Grecia y las regiones costeras turcas . Las principales islas mediterráneas (excepto las Baleares ), como Sicilia , Cerdeña , Córcega , Chipre , Malta y Creta , se ven gravemente afectadas en particular. [62] [63] Otros pueblos mediterráneos , así como los de las proximidades del Mediterráneo, también tienen altas tasas de incidencia, incluidas las personas de Asia occidental y el norte de África . En Pakistán , el 6% de la población total es portadora de beta-talasemia debido a la tasa más alta de matrimonios entre primos a nivel mundial. Los datos indican que el 15% de los chipriotas griegos y turcos son portadores de genes de beta-talasemia, mientras que el 10% de la población es portadora de genes de alfa-talasemia . [64]
El rasgo talasémico puede conferir un grado de protección contra la malaria , [65] que es o fue prevalente en las regiones donde el rasgo es común, confiriendo así una ventaja de supervivencia selectiva a los portadores (conocida como ventaja heterocigótica ), perpetuando así la mutación. En ese sentido, las diversas talasemias se parecen a otro trastorno genético que afecta a la hemoglobina, la anemia falciforme . [66]
El trastorno es más frecuente en ciertas etnias y grupos de edad. La beta talasemia es más frecuente en el "cinturón de talasemia", que incluye áreas del África subsahariana, el Mediterráneo que se extiende hasta Oriente Medio y el sudeste asiático. [8] Se cree que esta distribución geográfica se debe al estado de portador de beta-talasemia (beta talasemia menor) que confiere resistencia a la malaria. [8] En los Estados Unidos, la prevalencia de la talasemia es de aproximadamente 1 en 272.000 o 1.000 personas. Ha habido 4.000 casos hospitalizados en Inglaterra en 2002 y 9.233 episodios de consulta por talasemia. Los hombres representaron el 53% de los episodios de consulta hospitalaria y las mujeres representaron el 47%. La edad media de los pacientes es de 23 años, y solo el 1% de los consultores son mayores de 75 años y el 69% tienen entre 15 y 59 años. Se estima que el 1,5% de la población mundial es portadora y 40.000 niños afectados nacen con la enfermedad cada año. [8] La beta talasemia mayor suele ser mortal en la infancia si no se inician transfusiones de sangre inmediatamente. [67]