En química , una constante de disociación ácida (también conocida como constante de acidez o constante de ionización ácida ; denotada como ) es una medida cuantitativa de la fuerza de un ácido en solución . Es la constante de equilibrio de una reacción química .
conocido como disociación en el contexto de las reacciones ácido-base . La especie química HA es un ácido que se disocia en A − , llamada la base conjugada del ácido, y un ion hidrógeno , H + . [a] Se dice que el sistema está en equilibrio cuando las concentraciones de sus componentes no cambian con el tiempo, porque las reacciones tanto hacia adelante como hacia atrás ocurren al mismo ritmo. [1]
La constante de disociación se define por [b]
donde las cantidades entre corchetes representan las concentraciones molares de las especies en equilibrio. [c] [2] Por ejemplo, un ácido débil hipotético que tiene K a = 10 −5 , el valor de log K a es el exponente (−5), dando p K a = 5. Para el ácido acético , K a = 1,8 x 10 −5 , por lo que p K a es aproximadamente 5. Un K a más alto corresponde a un ácido más fuerte (un ácido que está más disociado en el equilibrio). La forma p K a se utiliza a menudo porque proporciona una escala logarítmica conveniente , donde un p K a más bajo corresponde a un ácido más fuerte.
La constante de disociación ácida de un ácido es una consecuencia directa de la termodinámica subyacente de la reacción de disociación; el valor de p K a es directamente proporcional al cambio de energía libre de Gibbs estándar para la reacción. El valor de p K a cambia con la temperatura y se puede entender cualitativamente con base en el principio de Le Châtelier : cuando la reacción es endotérmica , K a aumenta y p K a disminuye con el aumento de la temperatura; lo opuesto es cierto para las reacciones exotérmicas .
El valor de p K a también depende de la estructura molecular del ácido de muchas maneras. Por ejemplo, Pauling propuso dos reglas: una para p K a sucesivos de ácidos polipróticos (ver Ácidos polipróticos a continuación), y otra para estimar el p K a de oxiácidos basándose en el número de grupos =O y −OH (ver Factores que afectan los valores de pKa a continuación). Otros factores estructurales que influyen en la magnitud de la constante de disociación del ácido incluyen efectos inductivos , efectos mesoméricos y enlaces de hidrógeno . Las ecuaciones de tipo Hammett se han aplicado con frecuencia a la estimación de p K a . [3] [4]
El comportamiento cuantitativo de los ácidos y bases en solución sólo puede entenderse si se conocen sus valores p K a . En particular, el pH de una solución puede predecirse cuando se conocen la concentración analítica y los valores p K a de todos los ácidos y bases; a la inversa, es posible calcular la concentración de equilibrio de los ácidos y bases en solución cuando se conoce el pH. Estos cálculos encuentran aplicación en muchas áreas diferentes de la química, la biología, la medicina y la geología. Por ejemplo, muchos compuestos utilizados para la medicación son ácidos o bases débiles, y el conocimiento de los valores p K a , junto con el coeficiente de partición octanol-agua , puede utilizarse para estimar el grado en que el compuesto entra en el torrente sanguíneo. Las constantes de disociación ácida también son esenciales en la química acuática y la oceanografía química , donde la acidez del agua juega un papel fundamental. En los organismos vivos, la homeostasis ácido-base y la cinética enzimática dependen de los valores p K a de los muchos ácidos y bases presentes en la célula y en el cuerpo. En química, el conocimiento de los valores de p K a es necesario para la preparación de soluciones tampón y también es un prerrequisito para una comprensión cuantitativa de la interacción entre ácidos o bases e iones metálicos para formar complejos . Experimentalmente, los valores de p K a se pueden determinar mediante titulación potenciométrica (pH) , pero para valores de p K a menores de aproximadamente 2 o mayores de aproximadamente 11, pueden requerirse mediciones espectrofotométricas o de RMN debido a dificultades prácticas con las mediciones de pH.
Según la definición molecular original de Arrhenius , un ácido es una sustancia que se disocia en solución acuosa, liberando el ion hidrógeno H + (un protón): [5]
La constante de equilibrio para esta reacción de disociación se conoce como constante de disociación . El protón liberado se combina con una molécula de agua para dar un ion hidronio (u oxonio) H 3 O + (los protones desnudos no existen en solución), por lo que Arrhenius propuso posteriormente que la disociación debería escribirse como una reacción ácido-base :

Brønsted y Lowry generalizaron esto a una reacción de intercambio de protones: [6] [7] [8]
El ácido pierde un protón, dejando una base conjugada; el protón se transfiere a la base, creando un ácido conjugado. Para soluciones acuosas de un ácido HA, la base es agua; la base conjugada es A − y el ácido conjugado es el ion hidronio. La definición de Brønsted–Lowry se aplica a otros solventes, como el dimetilsulfóxido : el solvente S actúa como una base, aceptando un protón y formando el ácido conjugado SH + .
En la química de soluciones, es común utilizar H + como abreviatura del ion hidrógeno solvatado, independientemente del solvente. En solución acuosa, H + denota un ion hidronio solvatado en lugar de un protón. [9] [10]
La designación de un ácido o una base como "conjugado" depende del contexto. El ácido conjugado BH + de una base B se disocia según
que es el reverso del equilibrio
El ion hidróxido OH − , una base bien conocida, actúa aquí como base conjugada del agua ácida. Por lo tanto, los ácidos y las bases se consideran simplemente como donantes y aceptores de protones respectivamente.
Una definición más amplia de disociación ácida incluye la hidrólisis , en la que se producen protones mediante la división de las moléculas de agua. Por ejemplo, el ácido bórico ( B(OH) 3 ) produce H3O + como si fuera un donador de protones, [ 11] pero se ha confirmado mediante espectroscopia Raman que esto se debe al equilibrio de hidrólisis: [12]
De manera similar, la hidrólisis de iones metálicos hace que iones como [Al(H 2 O) 6 ] 3+ se comporten como ácidos débiles: [13]
Según la definición original de Lewis , un ácido es una sustancia que acepta un par de electrones para formar un enlace covalente coordinado . [14]
Una constante de disociación ácida es un ejemplo particular de una constante de equilibrio . La disociación de un ácido monoprótico , HA, en solución diluida se puede escribir como
La constante de equilibrio termodinámico se puede definir mediante [15 ]
donde representa la actividad , en equilibrio, de la especie química X. es adimensional ya que la actividad es adimensional. Las actividades de los productos de disociación se colocan en el numerador, las actividades de los reactivos se colocan en el denominador. Véase el coeficiente de actividad para una derivación de esta expresión.

Dado que la actividad es el producto de la concentración y el coeficiente de actividad ( γ ), la definición también podría escribirse como
donde representa la concentración de HA y es un cociente de coeficientes de actividad.
Para evitar las complicaciones que implica el uso de actividades, las constantes de disociación se determinan , cuando es posible, en un medio de alta fuerza iónica , es decir, en condiciones en las que se puede suponer que es siempre constante. [15] Por ejemplo, el medio podría ser una solución de nitrato de sodio 0,1 molar (M) o perclorato de potasio 3 M. Con esta suposición,
Se obtiene. Sin embargo, cabe señalar que todos los valores de constante de disociación publicados se refieren al medio iónico específico utilizado en su determinación y que se obtienen valores diferentes con diferentes condiciones, como se muestra para el ácido acético en la ilustración anterior. Cuando las constantes publicadas se refieren a una fuerza iónica distinta de la requerida para una aplicación particular, se pueden ajustar mediante la teoría de iones específicos (SIT) y otras teorías. [16]
Una constante de equilibrio acumulativa, denotada por está relacionada con el producto de constantes escalonadas, denotadas por Para un ácido dibásico, la relación entre las constantes escalonadas y las constantes generales es la siguiente
Obsérvese que en el contexto de la formación de complejos metal-ligando, las constantes de equilibrio para la formación de complejos metálicos se definen habitualmente como constantes de asociación . En ese caso, las constantes de equilibrio para la protonación del ligando también se definen como constantes de asociación. La numeración de las constantes de asociación es inversa a la numeración de las constantes de disociación; en este ejemplo
Cuando se discuten las propiedades de los ácidos, es habitual especificar las constantes de equilibrio como constantes de disociación ácida, denotadas por K a , con valores numéricos dados por el símbolo p K a .
Por otro lado, se utilizan constantes de asociación para las bases.
Sin embargo, los programas informáticos de uso general que se utilizan para derivar valores de constantes de equilibrio a partir de datos experimentales utilizan constantes de asociación tanto para ácidos como para bases. Debido a que las constantes de estabilidad para un complejo metal-ligando siempre se especifican como constantes de asociación, la protonación del ligando también debe especificarse como una reacción de asociación. [17] Las definiciones muestran que el valor de una constante de disociación de un ácido es el recíproco del valor de la constante de asociación correspondiente:
Notas
Todas las constantes de equilibrio varían con la temperatura según la ecuación de van 't Hoff [18]
es la constante de los gases y es la temperatura absoluta . Por lo tanto, para las reacciones exotérmicas , el cambio de entalpía estándar , , es negativo y K disminuye con la temperatura. Para las reacciones endotérmicas , es positivo y K aumenta con la temperatura.
El cambio de entalpía estándar para una reacción es en sí mismo una función de la temperatura, según la ley de termoquímica de Kirchhoff :
donde es el cambio de capacidad térmica a presión constante. En la práctica puede considerarse constante en un rango pequeño de temperaturas.
En la ecuación
K a parece tener dimensiones de concentración. Sin embargo, como , la constante de equilibrio, , no puede tener una dimensión física. Esta aparente paradoja se puede resolver de varias maneras.
Los procedimientos (1) y (2) dan valores numéricos idénticos para una constante de equilibrio. Además, dado que una concentración es simplemente proporcional a la fracción molar y la densidad :
y dado que la masa molar es una constante en soluciones diluidas, un valor constante de equilibrio determinado usando (3) será simplemente proporcional a los valores obtenidos con (1) y (2).
Es una práctica común en bioquímica citar un valor con una dimensión como, por ejemplo, " K a = 30 mM" para indicar la escala, milimolar (mM) o micromolar (μM) de los valores de concentración utilizados para su cálculo.
Un ácido se clasifica como "fuerte" cuando la concentración de sus especies no disociadas es demasiado baja para ser medida. [6] Cualquier ácido acuoso con un valor de p K a menor que 0 está casi completamente desprotonado y se considera un ácido fuerte . [20] Todos estos ácidos transfieren sus protones al agua y forman la especie catiónica del disolvente (H 3 O + en solución acuosa) de modo que todos tienen esencialmente la misma acidez, un fenómeno conocido como nivelación del disolvente . [21] [22] Se dice que están completamente disociados en solución acuosa porque la cantidad de ácido no disociado, en equilibrio con los productos de disociación, está por debajo del límite de detección . Del mismo modo, cualquier base acuosa con una constante de asociación p K b menor que aproximadamente 0, correspondiente a p K a mayor que aproximadamente 14, se nivela a OH − y se considera una base fuerte . [22]
El ácido nítrico , con un valor p K de alrededor de −1,7, se comporta como un ácido fuerte en soluciones acuosas con un pH mayor que 1. [23] A valores de pH más bajos se comporta como un ácido débil.
Los valores de p K a para ácidos fuertes se han estimado por medios teóricos. [24] Por ejemplo, el valor de p K a del HCl acuoso se ha estimado en −9,3.

Después de reordenar la expresión que define K a , y poner pH = −log 10 [H + ] , se obtiene [25]
Esta es la ecuación de Henderson-Hasselbalch , de la que se pueden extraer las siguientes conclusiones.
En el agua, los valores de p K a mensurables varían desde aproximadamente -2 para un ácido fuerte hasta aproximadamente 12 para un ácido muy débil (o base fuerte).
Se puede preparar una solución tampón de un pH deseado como una mezcla de un ácido débil y su base conjugada. En la práctica, la mezcla se puede crear disolviendo el ácido en agua y agregando la cantidad necesaria de ácido o base fuerte. Cuando se conocen el p K a y la concentración analítica del ácido, el grado de disociación y el pH de una solución de un ácido monoprótico se pueden calcular fácilmente utilizando una tabla ICE .

Un ácido poliprótico es un compuesto que puede perder más de un protón. Las constantes de disociación escalonada se definen para la pérdida de un solo protón. La constante para la disociación del primer protón se puede denotar como K a1 y las constantes para la disociación de protones sucesivos como K a2 , etc. El ácido fosfórico , H 3 PO 4 , es un ejemplo de ácido poliprótico, ya que puede perder tres protones.
Cuando la diferencia entre valores p K sucesivos es de aproximadamente cuatro o más, como en este ejemplo, cada especie puede considerarse un ácido por derecho propio; [27] De hecho, las sales de H
2correos−
4Puede cristalizarse a partir de una solución ajustando el pH a aproximadamente 5,5 y sales de HPO2−4Puede cristalizarse a partir de una solución ajustando el pH a aproximadamente 10. El diagrama de distribución de especies muestra que las concentraciones de los dos iones son máximas a pH 5,5 y 10.

Cuando la diferencia entre valores de p K sucesivos es menor que aproximadamente cuatro, existe una superposición entre el rango de pH de existencia de las especies en equilibrio. Cuanto menor sea la diferencia, mayor será la superposición. El caso del ácido cítrico se muestra a la derecha; las soluciones de ácido cítrico se tamponan en todo el rango de pH de 2,5 a 7,5.
Según la primera regla de Pauling, los valores p K sucesivos de un ácido dado aumentan (p K a2 > p K a1 ) . [28] Para los oxiácidos con más de un hidrógeno ionizable en el mismo átomo, los valores p K a a menudo aumentan alrededor de 5 unidades por cada protón eliminado, [29] [30] como en el ejemplo del ácido fosfórico mencionado anteriormente.
En la tabla anterior se puede observar que el segundo protón se elimina de una especie con carga negativa. Como el protón lleva una carga positiva, se necesita trabajo adicional para eliminarlo, por lo que p K a2 es mayor que p K a1 . p K a3 es mayor que p K a2 porque hay una mayor separación de cargas. Cuando se encuentra una excepción a la regla de Pauling, indica que también se está produciendo un cambio importante en la estructura. En el caso de VO+2(aq), el vanadio es octaédrico , de 6 coordenadas, mientras que el ácido vanádico es tetraédrico , de 4 coordenadas. Esto significa que se liberan cuatro "partículas" con la primera disociación, pero solo dos "partículas" con las otras disociaciones, lo que da como resultado una contribución de entropía mucho mayor al cambio de energía libre de Gibbs estándar para la primera reacción que para las otras.
Para las sustancias en solución, el punto isoeléctrico (p I ) se define como el pH en el que la suma, ponderada por el valor de la carga, de las concentraciones de las especies con carga positiva es igual a la suma ponderada de las concentraciones de las especies con carga negativa. En el caso de que haya una especie de cada tipo, el punto isoeléctrico se puede obtener directamente de los valores p K . Tomemos el ejemplo de la glicina , definida como AH. Hay dos equilibrios de disociación a considerar.
Sustituya la expresión para [AH] de la segunda ecuación en la primera ecuación.
En el punto isoeléctrico la concentración de las especies cargadas positivamente, AH+2, es igual a la concentración de la especie cargada negativamente, A − , por lo que
Por lo tanto, tomando cologaritmos , el pH viene dado por
Los valores de p I de los aminoácidos se enumeran en aminoácido proteinogénico . Cuando más de dos especies cargadas están en equilibrio entre sí, puede ser necesario un cálculo de especiación completo.
La constante de equilibrio K b para una base se define generalmente como la constante de asociación para la protonación de la base, B, para formar el ácido conjugado, HB + .
Usando un razonamiento similar al utilizado anteriormente
K b está relacionado con K a para el ácido conjugado. En el agua, la concentración del ion hidróxido , [OH − ] , está relacionada con la concentración del ion hidrógeno por K w = [H + ][OH − ] , por lo tanto
Sustituyendo la expresión para [OH − ] en la expresión para K b se obtiene
Cuando se determinan K a , K b y K w en las mismas condiciones de temperatura y fuerza iónica, se deduce, tomando cologaritmos , que p K b = p K w − p K a . En soluciones acuosas a 25 °C, p K w es 13,9965, [31] por lo que
con suficiente precisión para la mayoría de los propósitos prácticos. En efecto, no hay necesidad de definir p K b por separado de p K a , [32] pero se hace aquí ya que a menudo solo se pueden encontrar valores de p K b en la literatura más antigua.
Para un ion metálico hidrolizado, K b también se puede definir como una constante de disociación escalonada.
Este es el recíproco de una constante de asociación para la formación del complejo.
Debido a que la relación p K b = p K w − p K a se cumple solo en soluciones acuosas (aunque se aplican relaciones análogas para otros solventes anfóteros), las subdisciplinas de la química como la química orgánica que generalmente tratan con soluciones no acuosas generalmente no usan p K b como una medida de basicidad. En cambio, se cita el p K a del ácido conjugado, denotado por p K aH , cuando se necesita cuantificar la basicidad. Para la base B y su ácido conjugado BH + en equilibrio, esto se define como
Un valor más alto de p K aH corresponde a una base más fuerte. Por ejemplo, los valores p K aH (C 5 H 5 N) = 5,25 y p K aH ((CH 3 CH 2 ) 3 N) = 10,75 indican que (CH 3 CH 2 ) 3 N (trietilamina) es una base más fuerte que C 5 H 5 N (piridina).
Una sustancia anfótera es aquella que puede actuar como ácido o como base, dependiendo del pH. El agua (abajo) es anfótera. Otro ejemplo de molécula anfótera es el ion bicarbonato HCO−3Esa es la base conjugada de la molécula de ácido carbónico H2CO3 en el equilibrio.
pero también el ácido conjugado del ion carbonato CO2−3en (el reverso del) equilibrio
Los equilibrios del ácido carbónico son importantes para la homeostasis ácido-base en el cuerpo humano.
Un aminoácido también es anfótero con la complicación añadida de que la molécula neutra está sujeta a un equilibrio ácido-base interno en el que el grupo amino básico atrae y une el protón del grupo carboxilo ácido, formando un zwitterión .
A un pH inferior a aproximadamente 5, tanto el grupo carboxilato como el grupo amino están protonados. A medida que aumenta el pH, el ácido se disocia de acuerdo con
A un pH alto puede producirse una segunda disociación.
Por lo tanto, la molécula de aminoácido es anfótera porque puede estar protonada o desprotonada.
La molécula de agua puede ganar o perder un protón. Se dice que es anfiprótica . El equilibrio de ionización se puede escribir
where in aqueous solution H+ denotes a solvated proton. Often this is written as the hydronium ion H3O+, but this formula is not exact because in fact there is solvation by more than one water molecule and species such as H5O+2, H7O+3, and H9O+4 are also present.[33]
The equilibrium constant is given by
With solutions in which the solute concentrations are not very high, the concentration [H2O] can be assumed to be constant, regardless of solute(s); this expression may then be replaced by
The self-ionization constant of water, Kw, is thus just a special case of an acid dissociation constant. A logarithmic form analogous to pKa may also be defined
These data can be modelled by a parabola with
From this equation, pKw = 14 at 24.87 °C. At that temperature both hydrogen and hydroxide ions have a concentration of 10−7 M.
A solvent will be more likely to promote ionization of a dissolved acidic molecule in the following circumstances:[35]
pKa values of organic compounds are often obtained using the aprotic solvents dimethyl sulfoxide (DMSO)[35] and acetonitrile (ACN).[36]
DMSO is widely used as an alternative to water because it has a lower dielectric constant than water, and is less polar and so dissolves non-polar, hydrophobic substances more easily. It has a measurable pKa range of about 1 to 30. Acetonitrile is less basic than DMSO, and, so, in general, acids are weaker and bases are stronger in this solvent. Some pKa values at 25 °C for acetonitrile (ACN)[37][38][39] and dimethyl sulfoxide (DMSO).[40] are shown in the following tables. Values for water are included for comparison.
Ionization of acids is less in an acidic solvent than in water. For example, hydrogen chloride is a weak acid when dissolved in acetic acid. This is because acetic acid is a much weaker base than water.
Compare this reaction with what happens when acetic acid is dissolved in the more acidic solvent pure sulfuric acid:[41]

The unlikely geminal diol species CH3C(OH)+2 is stable in these environments. For aqueous solutions the pH scale is the most convenient acidity function.[42] Other acidity functions have been proposed for non-aqueous media, the most notable being the Hammett acidity function, H0, for superacid media and its modified version H− for superbasic media.[43]
In aprotic solvents, oligomers, such as the well-known acetic acid dimer, may be formed by hydrogen bonding. An acid may also form hydrogen bonds to its conjugate base. This process, known as homoconjugation, has the effect of enhancing the acidity of acids, lowering their effective pKa values, by stabilizing the conjugate base. Homoconjugation enhances the proton-donating power of toluenesulfonic acid in acetonitrile solution by a factor of nearly 800.[44]
In aqueous solutions, homoconjugation does not occur, because water forms stronger hydrogen bonds to the conjugate base than does the acid.

When a compound has limited solubility in water it is common practice (in the pharmaceutical industry, for example) to determine pKa values in a solvent mixture such as water/dioxane or water/methanol, in which the compound is more soluble.[46] In the example shown at the right, the pKa value rises steeply with increasing percentage of dioxane as the dielectric constant of the mixture is decreasing.
A pKa value obtained in a mixed solvent cannot be used directly for aqueous solutions. The reason for this is that when the solvent is in its standard state its activity is defined as one. For example, the standard state of water:dioxane mixture with 9:1 mixing ratio is precisely that solvent mixture, with no added solutes. To obtain the pKa value for use with aqueous solutions it has to be extrapolated to zero co-solvent concentration from values obtained from various co-solvent mixtures.
These facts are obscured by the omission of the solvent from the expression that is normally used to define pKa, but pKa values obtained in a given mixed solvent can be compared to each other, giving relative acid strengths. The same is true of pKa values obtained in a particular non-aqueous solvent such a DMSO.
A universal, solvent-independent, scale for acid dissociation constants has not been developed, since there is no known way to compare the standard states of two different solvents.
Pauling's second rule is that the value of the first pKa for acids of the formula XOm(OH)n depends primarily on the number of oxo groups m, and is approximately independent of the number of hydroxy groups n, and also of the central atom X. Approximate values of pKa are 8 for m = 0, 2 for m = 1, −3 for m = 2 and < −10 for m = 3.[28] Alternatively, various numerical formulas have been proposed including pKa = 8 − 5m (known as Bell's rule),[29][47] pKa = 7 − 5m,[30][48] or pKa = 9 − 7m.[29] The dependence on m correlates with the oxidation state of the central atom, X: the higher the oxidation state the stronger the oxyacid.
For example, pKa for HClO is 7.2, for HClO2 is 2.0, for HClO3 is −1 and HClO4 is a strong acid (pKa ≪ 0).[7] The increased acidity on adding an oxo group is due to stabilization of the conjugate base by delocalization of its negative charge over an additional oxygen atom.[47] This rule can help assign molecular structure: for example, phosphorous acid, having molecular formula H3PO3, has a pKa near 2, which suggested that the structure is HPO(OH)2, as later confirmed by NMR spectroscopy, and not P(OH)3, which would be expected to have a pKa near 8.[48]

Inductive effects and mesomeric effects affect the pKa values. A simple example is provided by the effect of replacing the hydrogen atoms in acetic acid by the more electronegative chlorine atom. The electron-withdrawing effect of the substituent makes ionisation easier, so successive pKa values decrease in the series 4.7, 2.8, 1.4, and 0.7 when 0, 1, 2, or 3 chlorine atoms are present.[49] The Hammett equation, provides a general expression for the effect of substituents.[50]
Ka is the dissociation constant of a substituted compound, K0
a is the dissociation constant when the substituent is hydrogen, ρ is a property of the unsubstituted compound and σ has a particular value for each substituent. A plot of log(Ka) against σ is a straight line with intercept log(K0
a) and slope ρ. This is an example of a linear free energy relationship as log(Ka) is proportional to the standard free energy change. Hammett originally[51] formulated the relationship with data from benzoic acid with different substituents in the ortho- and para- positions: some numerical values are in Hammett equation. This and other studies allowed substituents to be ordered according to their electron-withdrawing or electron-releasing power, and to distinguish between inductive and mesomeric effects.[52][53]
Alcohols do not normally behave as acids in water, but the presence of a double bond adjacent to the OH group can substantially decrease the pKa by the mechanism of keto–enol tautomerism. Ascorbic acid is an example of this effect. The diketone 2,4-pentanedione (acetylacetone) is also a weak acid because of the keto–enol equilibrium. In aromatic compounds, such as phenol, which have an OH substituent, conjugation with the aromatic ring as a whole greatly increases the stability of the deprotonated form.
Structural effects can also be important. The difference between fumaric acid and maleic acid is a classic example. Fumaric acid is (E)-1,4-but-2-enedioic acid, a trans isomer, whereas maleic acid is the corresponding cis isomer, i.e. (Z)-1,4-but-2-enedioic acid (see cis-trans isomerism). Fumaric acid has pKa values of approximately 3.0 and 4.5. By contrast, maleic acid has pKa values of approximately 1.5 and 6.5. The reason for this large difference is that when one proton is removed from the cis isomer (maleic acid) a strong intramolecular hydrogen bond is formed with the nearby remaining carboxyl group. This favors the formation of the maleate H+, and it opposes the removal of the second proton from that species. In the trans isomer, the two carboxyl groups are always far apart, so hydrogen bonding is not observed.[54]

Proton sponge, 1,8-bis(dimethylamino)naphthalene, has a pKa value of 12.1. It is one of the strongest amine bases known. The high basicity is attributed to the relief of strain upon protonation and strong internal hydrogen bonding.[55][56]
Effects of the solvent and solvation should be mentioned also in this section. It turns out, these influences are more subtle than that of a dielectric medium mentioned above. For example, the expected (by electronic effects of methyl substituents) and observed in gas phase order of basicity of methylamines, Me3N > Me2NH > MeNH2 > NH3, is changed by water to Me2NH > MeNH2 > Me3N > NH3. Neutral methylamine molecules are hydrogen-bonded to water molecules mainly through one acceptor, N–HOH, interaction and only occasionally just one more donor bond, NH–OH2. Hence, methylamines are stabilized to about the same extent by hydration, regardless of the number of methyl groups. In stark contrast, corresponding methylammonium cations always utilize all the available protons for donor NH–OH2 bonding. Relative stabilization of methylammonium ions thus decreases with the number of methyl groups explaining the order of water basicity of methylamines.[4]
An equilibrium constant is related to the standard Gibbs energy change for the reaction, so for an acid dissociation constant
R is the gas constant and T is the absolute temperature. Note that pKa = −log(Ka) and 2.303 ≈ ln(10). At 25 °C, ΔG⊖ in kJ·mol−1 ≈ 5.708 pKa (1 kJ·mol−1 = 1000 joules per mole). Free energy is made up of an enthalpy term and an entropy term.[11]
The standard enthalpy change can be determined by calorimetry or by using the van 't Hoff equation, though the calorimetric method is preferable. When both the standard enthalpy change and acid dissociation constant have been determined, the standard entropy change is easily calculated from the equation above. In the following table, the entropy terms are calculated from the experimental values of pKa and ΔH⊖. The data were critically selected and refer to 25 °C and zero ionic strength, in water.[11]
The first point to note is that, when pKa is positive, the standard free energy change for the dissociation reaction is also positive. Second, some reactions are exothermic and some are endothermic, but, when ΔH⊖ is negative TΔS⊖ is the dominant factor, which determines that ΔG⊖ is positive. Last, the entropy contribution is always unfavourable (ΔS⊖ < 0) in these reactions. Ions in aqueous solution tend to orient the surrounding water molecules, which orders the solution and decreases the entropy. The contribution of an ion to the entropy is the partial molar entropy which is often negative, especially for small or highly charged ions.[57] The ionization of a neutral acid involves formation of two ions so that the entropy decreases (ΔS⊖ < 0). On the second ionization of the same acid, there are now three ions and the anion has a charge, so the entropy again decreases.
Note that the standard free energy change for the reaction is for the changes from the reactants in their standard states to the products in their standard states. The free energy change at equilibrium is zero since the chemical potentials of reactants and products are equal at equilibrium.

The experimental determination of pKa values is commonly performed by means of titrations, in a medium of high ionic strength and at constant temperature.[58] A typical procedure would be as follows. A solution of the compound in the medium is acidified with a strong acid to the point where the compound is fully protonated. The solution is then titrated with a strong base until all the protons have been removed. At each point in the titration pH is measured using a glass electrode and a pH meter. The equilibrium constants are found by fitting calculated pH values to the observed values, using the method of least squares.[59]
The total volume of added strong base should be small compared to the initial volume of titrand solution in order to keep the ionic strength nearly constant. This will ensure that pKa remains invariant during the titration.
A calculated titration curve for oxalic acid is shown at the right. Oxalic acid has pKa values of 1.27 and 4.27. Therefore, the buffer regions will be centered at about pH 1.3 and pH 4.3. The buffer regions carry the information necessary to get the pKa values as the concentrations of acid and conjugate base change along a buffer region.
Between the two buffer regions there is an end-point, or equivalence point, at about pH 3. This end-point is not sharp and is typical of a diprotic acid whose buffer regions overlap by a small amount: pKa2 − pKa1 is about three in this example. (If the difference in pK values were about two or less, the end-point would not be noticeable.) The second end-point begins at about pH 6.3 and is sharp. This indicates that all the protons have been removed. When this is so, the solution is not buffered and the pH rises steeply on addition of a small amount of strong base. However, the pH does not continue to rise indefinitely. A new buffer region begins at about pH 11 (pKw − 3), which is where self-ionization of water becomes important.
It is very difficult to measure pH values of less than two in aqueous solution with a glass electrode, because the Nernst equation breaks down at such low pH values. To determine pK values of less than about 2 or more than about 11 spectrophotometric[60][61] or NMR[62][63] measurements may be used instead of, or combined with, pH measurements.
When the glass electrode cannot be employed, as with non-aqueous solutions, spectrophotometric methods are frequently used.[38] These may involve absorbance or fluorescence measurements. In both cases the measured quantity is assumed to be proportional to the sum of contributions from each photo-active species; with absorbance measurements the Beer–Lambert law is assumed to apply.
Isothermal titration calorimetry (ITC) may be used to determine both a pK value and the corresponding standard enthalpy for acid dissociation.[64] Software to perform the calculations is supplied by the instrument manufacturers for simple systems.
Aqueous solutions with normal water cannot be used for 1H NMR measurements but heavy water, D2O, must be used instead. 13C NMR data, however, can be used with normal water and 1H NMR spectra can be used with non-aqueous media. The quantities measured with NMR are time-averaged chemical shifts, as proton exchange is fast on the NMR time-scale. Other chemical shifts, such as those of 31P can be measured.

For some polyprotic acids, dissociation (or association) occurs at more than one nonequivalent site,[4] and the observed macroscopic equilibrium constant, or macro-constant, is a combination of micro-constants involving distinct species. When one reactant forms two products in parallel, the macro-constant is a sum of two micro-constants, This is true for example for the deprotonation of the amino acid cysteine, which exists in solution as a neutral zwitterion HS−CH2−CH(NH+3)−COO−. The two micro-constants represent deprotonation either at sulphur or at nitrogen, and the macro-constant sum here is the acid dissociation constant [65]
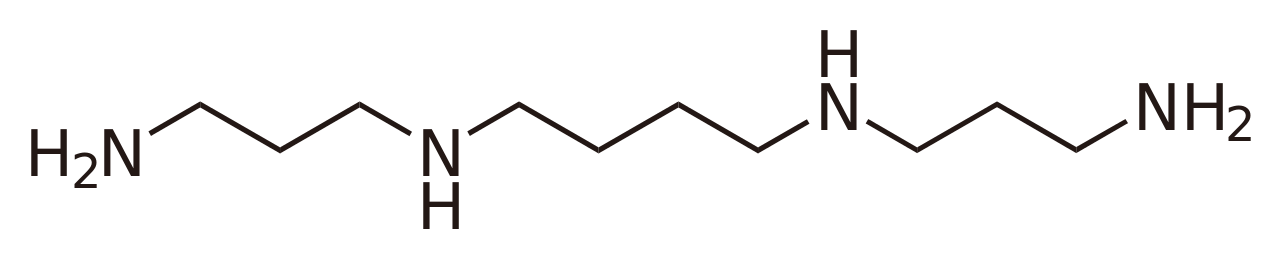
Similarly, a base such as spermine has more than one site where protonation can occur. For example, mono-protonation can occur at a terminal −NH2 group or at internal −NH− groups. The Kb values for dissociation of spermine protonated at one or other of the sites are examples of micro-constants. They cannot be determined directly by means of pH, absorbance, fluorescence or NMR measurements; a measured Kb value is the sum of the K values for the micro-reactions.
Nevertheless, the site of protonation is very important for biological function, so mathematical methods have been developed for the determination of micro-constants.[66]
When two reactants form a single product in parallel, the macro-constant [65] For example, the abovementioned equilibrium for spermine may be considered in terms of Ka values of two tautomeric conjugate acids, with macro-constant In this case This is equivalent to the preceding expression since is proportional to
When a reactant undergoes two reactions in series, the macro-constant for the combined reaction is the product of the micro-constant for the two steps. For example, the abovementioned cysteine zwitterion can lose two protons, one from sulphur and one from nitrogen, and the overall macro-constant for losing two protons is the product of two dissociation constants [65] This can also be written in terms of logarithmic constants as
A knowledge of pKa values is important for the quantitative treatment of systems involving acid–base equilibria in solution. Many applications exist in biochemistry; for example, the pKa values of proteins and amino acid side chains are of major importance for the activity of enzymes and the stability of proteins.[67] Protein pKa values cannot always be measured directly, but may be calculated using theoretical methods. Buffer solutions are used extensively to provide solutions at or near the physiological pH for the study of biochemical reactions;[68] the design of these solutions depends on a knowledge of the pKa values of their components. Important buffer solutions include MOPS, which provides a solution with pH 7.2, and tricine, which is used in gel electrophoresis.[69][70] Buffering is an essential part of acid base physiology including acid–base homeostasis,[71] and is key to understanding disorders such as acid–base disorder.[72][73][74] The isoelectric point of a given molecule is a function of its pK values, so different molecules have different isoelectric points. This permits a technique called isoelectric focusing,[75] which is used for separation of proteins by 2-D gel polyacrylamide gel electrophoresis.
Buffer solutions also play a key role in analytical chemistry. They are used whenever there is a need to fix the pH of a solution at a particular value. Compared with an aqueous solution, the pH of a buffer solution is relatively insensitive to the addition of a small amount of strong acid or strong base. The buffer capacity[76] of a simple buffer solution is largest when pH = pKa. In acid–base extraction, the efficiency of extraction of a compound into an organic phase, such as an ether, can be optimised by adjusting the pH of the aqueous phase using an appropriate buffer. At the optimum pH, the concentration of the electrically neutral species is maximised; such a species is more soluble in organic solvents having a low dielectric constant than it is in water. This technique is used for the purification of weak acids and bases.[77]
A pH indicator is a weak acid or weak base that changes colour in the transition pH range, which is approximately pKa ± 1. The design of a universal indicator requires a mixture of indicators whose adjacent pKa values differ by about two, so that their transition pH ranges just overlap.
In pharmacology, ionization of a compound alters its physical behaviour and macro properties such as solubility and lipophilicity, log p). For example, ionization of any compound will increase the solubility in water, but decrease the lipophilicity. This is exploited in drug development to increase the concentration of a compound in the blood by adjusting the pKa of an ionizable group.[78]
Knowledge of pKa values is important for the understanding of coordination complexes, which are formed by the interaction of a metal ion, Mm+, acting as a Lewis acid, with a ligand, L, acting as a Lewis base. However, the ligand may also undergo protonation reactions, so the formation of a complex in aqueous solution could be represented symbolically by the reaction
To determine the equilibrium constant for this reaction, in which the ligand loses a proton, the pKa of the protonated ligand must be known. In practice, the ligand may be polyprotic; for example EDTA4− can accept four protons; in that case, all pKa values must be known. In addition, the metal ion is subject to hydrolysis, that is, it behaves as a weak acid, so the pK values for the hydrolysis reactions must also be known.[79]
Assessing the hazard associated with an acid or base may require a knowledge of pKa values.[80] For example, hydrogen cyanide is a very toxic gas, because the cyanide ion inhibits the iron-containing enzyme cytochrome c oxidase. Hydrogen cyanide is a weak acid in aqueous solution with a pKa of about 9. In strongly alkaline solutions, above pH 11, say, it follows that sodium cyanide is "fully dissociated" so the hazard due to the hydrogen cyanide gas is much reduced. An acidic solution, on the other hand, is very hazardous because all the cyanide is in its acid form. Ingestion of cyanide by mouth is potentially fatal, independently of pH, because of the reaction with cytochrome c oxidase.
In environmental science acid–base equilibria are important for lakes[81] and rivers;[82][83] for example, humic acids are important components of natural waters. Another example occurs in chemical oceanography:[84] in order to quantify the solubility of iron(III) in seawater at various salinities, the pKa values for the formation of the iron(III) hydrolysis products Fe(OH)2+, Fe(OH)+2 and Fe(OH)3 were determined, along with the solubility product of iron hydroxide.[85]
There are multiple techniques to determine the pKa of a chemical, leading to some discrepancies between different sources. Well measured values are typically within 0.1 units of each other. Data presented here were taken at 25 °C in water.[7][86] More values can be found in the Thermodynamics section, above. A table of pKa of carbon acids, measured in DMSO, can be found on the page on carbanions.
Are you wondering... How using activities makes the equilibrium constant dimensionless?


![{\displaystyle K_{\text{a}}=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7cdd9efda0e3a32060020b5c9e5b2c78981b2a93)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}K_{\text{a}}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{\ce {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d7af05bf129db2f9bc618fe809660b6e4ff8dce9)







![{\displaystyle {\ce {[Al(H2O)6]^3+ + H2O <=> [Al(H2O)5(OH)]^2+ + H3O+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b1c60923d504a87f8bbd22293ac8eaad8341ea41)



![{\displaystyle K^{\ominus }={{\frac {[{\ce {A^-}}][{\ce {H+}}]}{{\ce {[HA]}}}}\Gamma },\quad \Gamma ={\frac {\gamma _{{\ce {A^-}}}\ \gamma _{{\ce {H+}}}}{\gamma _{{\ce {HA} }}\ }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6e9373db7091aeb4f51a26757a677b420f0a8418)
![{\displaystyle [{\text{HA}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3cfe8305c0735d25de8cef20edf09ef5144d700a)

![{\displaystyle K_{\text{a}}={\frac {K^{\ominus }}{\Gamma }}=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2a5a59c740de89347ec4c96d982292fc05c64b2f)
![{\displaystyle \mathrm {p} K_{{\ce {a}}}=-\log _{10}{\frac {[{\ce {A^-}}][{\ce {H^+} }]}{[{\ce {[HA}}]}}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{ \ce{H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bed5fbab82167a42994a6d735931d08b06f1e7a5)



![{\displaystyle \beta _{2}={\frac {{\ce {[H2A]}}}{[{\ce {A^2-}}][{\ce {H+}}]^{2} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/08598ffd39aa7af9e4d7ca73764ada00fdc0882f)


![{\displaystyle K_{\text{disoc}}={\frac {{\ce {[A- ][H+]}}}{{\ce {[HA]}}}}:\mathrm {p} K_{\text{a}}=-\log K_{\text{disoc}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c115ab88c5f847b2fe5c3250d9e5c9134d125080)
![{\displaystyle K_{\text{assoc}}={\frac {{\ce {[HA]}}}{{\ce {[A- ][H+]}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e353beeaa76919ab646b6969e6d30f7e01fe7afa)










![{\displaystyle K_{\mathrm {a}}=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/441ece0dee32e0a14fe14d4b1678785804486a92)







![{\displaystyle \mathrm {pH} =\mathrm {p} K_{\text{a}}+\log \mathrm {\frac {[A^{-}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/25e874f2b8ea8e4127605788c356393cfd7fff37)
![{\displaystyle {\ce {AH2+<=>AH~+H+\qquad [AH][H+]={\mathit {K}}_{1}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/359f1d34ddc5ac4b4cecf45e11539bc14462e98f)
![{\displaystyle {\ce {AH<=>A^{-}~+H+\qquad [A^{-}][H+]={\mathit {K}}_{2}[AH]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95c1af1bef047675675659473e3886aefff9fa79)
![{\displaystyle {\ce {[A^{-}][H+]^{2}={\mathit {K}}_{1}{\mathit {K}}_{2}[AH2+]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/473772e02e925c83975026ce31b43a0a2dc4b1cc)
![{\displaystyle [{\ce {H+}}]^{2}=K_{1}K_{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3dfa9985f2d6769f35f98f98153ac6cabd9c011b)


![{\displaystyle {\begin{aligned}K_{\text{b}}&=\mathrm {\frac {[HB^{+}][OH^{-}]}{[B]}} \\\mathrm {p} K_{\text{b}}&=-\log _{10}\left(K_{\text{b}}\right)\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5dea1aac629a595476e18c042a8f4365a50f0efc)
![{\displaystyle \mathrm {[OH^{-}]} ={\frac {K_{\mathrm {w} }}{\mathrm {[H^{+}]} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ab7f583da9f8b50145990ffa4342919930edfa16)
![{\displaystyle K_{\text{b}}={\frac {[\mathrm {HB^{+}} ]K_{\text{w}}}{\mathrm {[B][H^{+}]} }}={\frac {K_{\text{w}}}{K_{\text{a}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/921c3abd37a1c5c00c31831509d3b090394c0d47)


![{\displaystyle K_{b}}={\frac {[M_{p}({\ce {OH}})_{q-1}^{+}][{\ce {OH-}}]}{[M_{p}({\ce {OH}})_{q}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8391968e700e57f56f6041dd839f0fdb5e93780e)
![{\displaystyle \mathrm {p} K_{\mathrm {a} }(\mathrm {B} )=\mathrm {p} K_{\mathrm {a} }({\ce {BH+}})=-\log _{10}{\Grande (}{\frac {[{\ce {B}}][{\ce {H+}}]}{[{\ce {BH+}}]}}{\Grande )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b76149bc3dbc3d0375d6355bdf2342394a568776)






![{\displaystyle K_{\text{a}}=\mathrm {\frac {[H^{+}][OH^{-}]}{[H_{2}O]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/cdc540ad193c8f1661c1897698be93153fc5fb84)
![{\displaystyle K_{\text{w}}=[\mathrm {H} ^{+}][\mathrm {OH} ^{-}]\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0039f77db244ea2f6d03d3475dc7a232a8ccb16)
















![{\displaystyle [{\ce {M(H2O)_{\mathit {n}}}}]^{m+}+{\ce {LH<=>}}\ [{\ce {M(H2O)}}_{n-1}{\ce {L}}]^{(m-1)+}+{\ce {H3O+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/696ff04eb241a2f6a358b8dd1b9c373ea3a8c91d)