Síndrome de Klinefelter
El síndrome de Klinefelter (SK) o 47,XXY es la caracterización clínica de una mutación cromosómica que afecta a varones y que incluye, entre otras manifestaciones, hipogonadismo hipergonadotrópico, ginecomastia, dificultades en el aprendizaje e infertilidad.
La principal característica es la infertilidad, así como escroto hipoplásico y en ocasiones criptorquidia o micropene.
[8][9] En la edad adulta presentan una talla elevada y tendencia al sobrepeso; los individuos afectados tienen brazos y piernas largos.Pueden manifestar ginecomastia unilateral o bilateral, la cual se caracteriza por el desarrollo de pechos en el hombre (tejido mamario agrandado).
Es particularmente difícil para estos pacientes interpretar los sentimientos de otras personas solamente al escuchar lo que les dicen.
Sin embargo, presentan mayor dificultad al realizar trabajos que impliquen mucha lectura y escritura.
Estos pacientes también presentan trastornos emocionales como ansiedad, depresión, y tendencia al abuso de sustancias.
Esos pacientes son más propensos a enfermedades cardiovasculares dada la prevalencia en anormalidades metabólicas incluyendo dislipidemia y diabetes mellitus tipo 2.
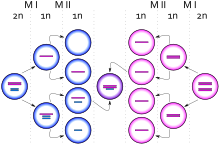
[10][13][14][15] La anomalía cromosómica puede originarse también por un error durante las divisiones mitóticas del cigoto, produciendo así los casos de mosaicismo.
[8][13][14] Otro mecanismo para retener un cromosoma extra es mediante la no disyunción en la meiosis II en el óvulo.
Esto produce un óvulo XX que posteriormente será fertilizado por un espermatozoide normal, dando un descendiente XXY (Klinefelter).
En estos casos la sintomatología puede ser más pronunciada que en los pacientes con síndrome de Klinefelter típico (47,XXY).
Por lo general, se presentan convulsiones en la infancia, estrabismo, problemas intestinales como estreñimiento, e infecciones en los oídos.
[7][17][18] Los varones con este cariotipo llegan a ser muy altos, con piernas muy largas; lo que puede resultar en úlceras y várices en las mismas.
[19] En el ámbito conductual, son extremadamente tímidos, aunque pueden llegar a ser agresivos e impulsivos.
Tienen la tendencia a tener mayores problemas de hiperactividad, agresión y depresión en comparación con los varones 46,XY o incluso 47,XXY.
[20] Quienes tienen este cariotipo presentan hipertelorismo ocular, un puente nasal plano, y son de baja estatura.
Los huesos de sus brazos pueden estar conectados de manera inusual, a lo que se le conoce como sinostosis radioulnar, o presentar clinodactilia, es decir que el dedo meñique se encuentra curvado hacia adentro.
[17] Su coeficiente intelectual estará entre los 40 y 60 puntos, lo que se hará evidente al tener un severo retraso en el habla.
Consistente con ello, son sujetos inmaduros, considerados incluso pasivos, cooperativos y no agresivos.
Sin embargo, la respuesta a la gonadoliberina (LHRH, hormona hipotalámica liberadora de gonadotropinas) es normal en ambos grupos.
Estos cambios histológicos son muy característicos del síndrome, y los cuales originan disminución en el volumen testicular así como aumento de su consistencia.
[34][35][36] En el caso en que no se puedan obtener espermatozoides funcionales a partir del semen o TESE, no hay posibilidad de producir hijos biológicos; esto debido a que el tratamiento de testosterona suprimiría la espermatogénesis.
Si la terapia de testosterona ya se ha iniciado, pero el paciente se desea todavía una TESE, la terapia de testosterona debe pausarse durante al menos 6 meses para que la espermatogénesis puede recuperarse.
No existe evidencia que indique de dicho proceso se repita en una familia en particular.
Tres años más tarde, en 1959, investigadores del Western General Hospital en Edimburgo, Escocia identificaron que el cariotipo de un sujeto con la enfermedad era 47,XXY.