Biopython
El proyecto 'Biopython' es el nombre que recibe una serie de aplicaciones y programas informáticos pensados para cuantificar y hacer cálculos con datos biológicos, programados por una comunidad internacional.
Entre los primeros desarrolladores se cuentan Jeff Chang, Andrew Dalke and Brad Chapman, aunque más de 100 colaboradores han contribuido al proyecto hasta la fecha.
[2] Un concepto central de Biopython es la secuencia biológica, que aparece representada por la clase Seq.
Además, es capaz incluir diferentes métodos específicos para secuencias determinadas y permite elegir el alfabeto biológico que se quiere utilizar.
La clase SeqRecord describe secuencias genéticas, junto con información como su nombre, descripción y características bajo la forma de objetos SeqFeature.
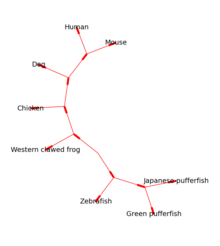
[5] Los árboles enraizados pueden ser dibujados en formato ASCII o usando matplotlib (ver Figura 1), y la biblioteca Graphviz puede ser utilizada para dibujar presentaciones sin enraizar (ver Figura 2).
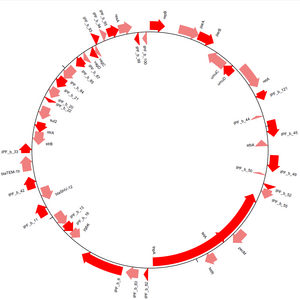
[7] Las secuencias pueden ser dibujadas en una forma linear o circular (ver Figura 3), y varios formatos de salida son posibles, incluyendo pdfs y pngs.
También se pueden dibujar enlaces entre las diferentes pistas, lo cual permite comparar múltiples secuencias en un solo diagrama.
Estos controladores incluyen BLAST, Clustal, PhyML, EMBOSS y SAMtools.