En química orgánica , la síntesis de Paal-Knorr es una reacción que se utiliza para sintetizar furanos , pirroles o tiofenos sustituidos a partir de 1,4-dicetonas . Es un método sintético valioso para obtener furanos y pirroles sustituidos, que son componentes estructurales comunes de muchos productos naturales. Los químicos alemanes Carl Paal y Ludwig Knorr lo informaron inicialmente de forma independiente en 1884 como un método para la preparación de furanos, y se ha adaptado para pirroles y tiofenos. [1] [2] Aunque la síntesis de Paal-Knorr ha tenido un uso generalizado, el mecanismo no se entendió por completo hasta que fue dilucidado por V. Amarnath et al. en la década de 1990. [3] [4]
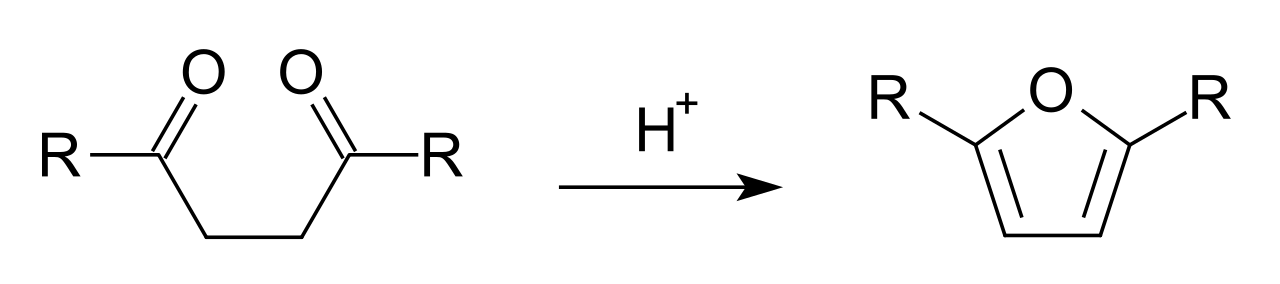
La síntesis de furano requiere un catalizador ácido: [5]
En la síntesis del pirrol participa una amina primaria :

y en el caso del tiofeno , por ejemplo, el compuesto pentasulfuro de fósforo :

La síntesis de furano catalizada por ácido se produce por protonación de un carbonilo que es atacado por el enol en formación del otro carbonilo. La deshidratación del hemiacetal da lugar al furano resultante. [6]

El mecanismo de la síntesis de furanos de Paal-Knorr fue dilucidado en 1995 por V. Amarnath et al . [3] El trabajo de Amarnath mostró que los diastereómeros de las dionas 3,4-disustituidas-2,5-hexano reaccionan a diferentes velocidades. En el mecanismo comúnmente aceptado, estas dionas pasarían por un intermediario enólico común, lo que significa que los isómeros meso y d,l -racémicos se ciclarían a la misma velocidad que se forman a partir de un intermediario común. La implicación de una reacción diferente es que la ciclización debe ocurrir en un paso concertado con la formación de enol. Por lo tanto, se propuso que el mecanismo ocurriera mediante el ataque del carbonilo protonado con el enol en formación. Amarnath también descubrió que la diona que no había reaccionado no había experimentado isomerización conformacional, lo que también indicaba que un enol no era un intermediario.
El mecanismo de síntesis del pirrol fue investigado por V. Amarnath et al. en 1991. [4] Su trabajo sugiere que el carbonilo protonado es atacado por la amina para formar el hemiaminal. La amina ataca al otro carbonilo para formar un derivado 2,5-dihidroxitetrahidropirrol que sufre deshidratación para dar el pirrol sustituido correspondiente. [7]

La reacción se lleva a cabo normalmente en condiciones próticas o ácidas de Lewis, con una amina primaria. El uso de hidróxido de amonio o acetato de amonio (como informó Paal) da el pirrol N-no sustituido.
La síntesis de tiofeno se logra mediante un mecanismo muy similar a la síntesis de furano. La dicetona inicial se convierte en una tiocetona con un agente sulfurante, que luego experimenta el mismo mecanismo que la síntesis de furano. [8]

La mayoría de los agentes de sulfuración son deshidratantes fuertes e impulsan la finalización de la reacción. Los primeros postulados sobre el mecanismo de la síntesis de furanos de Paal-Knorr sugirieron que el tiofeno se lograba por sulfuración del producto furano. Campaigne y Foye demostraron que el tratamiento de furanos aislados de la síntesis de furanos de Paal-Knorr con pentasulfuro de fósforo dio resultados inconsistentes con el tratamiento de 1,4-dicarbonilos con pentasulfuro de fósforo, lo que descartó la sulfuración de un mecanismo de furano y sugiere que la reacción procede a través de la sulfuración de un dicarbonilo, produciendo una tiocetona . [8]

La reacción de Paal-Knorr es bastante versátil. En todas las síntesis, casi todos los dicarbonilos pueden convertirse en su heterociclo correspondiente. R2 y R5 pueden ser H, arilo o alquilo. R3 y R4 pueden ser H, arilo, alquilo o un éster. En la síntesis de pirrol (X = N), R1 puede ser H, arilo, alquilo, amino o hidroxilo. [9]
Se pueden utilizar diversas condiciones para llevar a cabo estas reacciones, la mayoría de las cuales son suaves. La síntesis de furano de Paal-Knorr se lleva a cabo normalmente en condiciones ácidas acuosas con ácidos próticos como ácido sulfúrico o clorhídrico acuoso , o en condiciones anhidras con un ácido de Lewis o un agente deshidratante. Los agentes deshidratantes comunes incluyen pentóxido de fósforo , anhídridos o cloruro de cinc. La síntesis de pirrol requiere una amina primaria en condiciones similares, o se puede utilizar amoníaco (o precursores de amoníaco). La síntesis de un tiofeno requiere un agente sulfurante que normalmente es un deshidratante suficiente, como pentasulfuro de fósforo , reactivo de Lawesson o sulfuro de hidrógeno .
Tradicionalmente, la reacción de Paal-Knorr ha estado limitada en su alcance por la disponibilidad de 1,4-dicetonas como precursores sintéticos. Los métodos químicos actuales han ampliado enormemente la accesibilidad de estos reactivos, y las variaciones de Paal-Knorr ahora permiten el uso de diferentes precursores. La reacción de Paal-Knorr también se consideró limitada por las duras condiciones de reacción, como el calentamiento prolongado en ácido, que puede degradar las funcionalidades sensibles en muchos precursores potenciales de furano. Los métodos actuales permiten condiciones más suaves que pueden evitar el calor por completo, incluidas las ciclizaciones catalizadas por microondas .
Se pueden utilizar varios sustitutos del 1,4-dicarbonilo en lugar de un 1,4-dicarbonilo. Si bien estos sustitutos tienen estructuras diferentes a las del 1,4-dicarbonilo, sus reacciones se producen a través de mecanismos muy similares a los del Paal-Knorr.
Se sabe que los β-epoxicarbonilos se ciclan para formar furanos. Este procedimiento puede utilizar los carbonilos β-γ-insaturados como materiales de partida, que pueden epoxidarse. El epoxicarbonilo resultante puede ciclarse para formar un furano en condiciones ácidas o básicas. [10]

También se han utilizado sistemas de 1,4-diol-2-ino para realizar la química de Paal-Knorr. Utilizando paladio, un 1,4-diol-2-ino se puede isomerizar a la 1,4-dicetona correspondiente in situ y luego deshidratarlo al furano correspondiente utilizando un agente deshidratante. [11]

La importancia de esta variación radica en el hecho de que aumenta el alcance del método Paal-Knorr al aprovechar la riqueza de la química del acetileno que existe, específicamente la para la generación de alcoholes propargílicos.
Los acetales también han demostrado ser útiles como materiales de partida para la reacción de Paal-Knorr. Una cetona con un acetal a 3 enlaces de distancia puede convertirse en el heterociclo correspondiente en exactamente las mismas condiciones que una 1,4-dicetona.
Otra variación ha sido la introducción de radiación de microondas para mejorar la reacción de Paal-Knorr. Las condiciones tradicionales de Paal-Knorr implicaban el calentamiento prolongado de ácidos fuertes para impulsar la deshidratación, que se producía durante un período de varias horas. Se ha demostrado que las reacciones de Paal-Knorr asistidas por microondas se producen en escalas de tiempo medidas en minutos y en matraces abiertos a temperatura ambiente. [12]
La síntesis de pirrol de Knorr , informada por Knorr en 1884, es la síntesis de un pirrol sustituido a partir de una aminocetona y una cetona. [13]
Knorr también informó sobre la síntesis de pirazoles a partir de 1,3-dicarbonilos e hidrazinas , hidrazidas o semibicarbazidas. Esta síntesis se produce a través de un mecanismo de condensación similar al de Paal-Knorr, sin embargo, si se utiliza una hidrazina sustituida, se obtiene una mezcla de regioisómeros en la que el heteroátomo sustituido está junto al sustituyente R1 o al sustituyente R3. [14]

En 2000, BM Trost et al. informaron sobre una síntesis formal del antibiótico roseophilina. La ruta de Trost hacia el núcleo macrocíclico de la roseophilina, como la de otros, se basó en una síntesis de pirrol de Paal-Knorr para obtener el pirrol fusionado. [15] El calentamiento de la 1,4-dicetona con acetato de amonio en metanol con ácido alcanforsulfónico y tamices moleculares de 4 angstroms proporcionó el pirrol sin N-sustitución. Se descubrió que este pirrol era inestable y, como tal, se trató con cloruro de trimetilsilil etoxi metoxi (SEM-Cl) para protegerlo antes del aislamiento.

En 1982, H. Hart et al. informaron sobre la síntesis de un macrociclo que contenía anillos de furano fusionados utilizando una síntesis de furano de Paal-Knorr. [16] Se descubrió que el reflujo de ácido para -toluenosulfónico en benceno deshidrataba las 1,4-dicetonas a sus respectivos furanos para lograr los desafiantes furanos fusionados macrocíclicos.
