El programa de nuevos fármacos en investigación ( IND ) de la Administración de Alimentos y Medicamentos de los Estados Unidos es el medio por el cual una compañía farmacéutica obtiene permiso para iniciar ensayos clínicos en humanos y enviar un fármaco experimental a través de las fronteras estatales (generalmente a investigadores clínicos) antes de que se haya aprobado una solicitud de comercialización del fármaco. Las regulaciones se encuentran principalmente en 21 CFR 312. Se siguen procedimientos similares en la Unión Europea, Japón y Canadá debido a los esfuerzos de armonización regulatoria del Consejo Internacional para la Armonización . [1]
Tipos
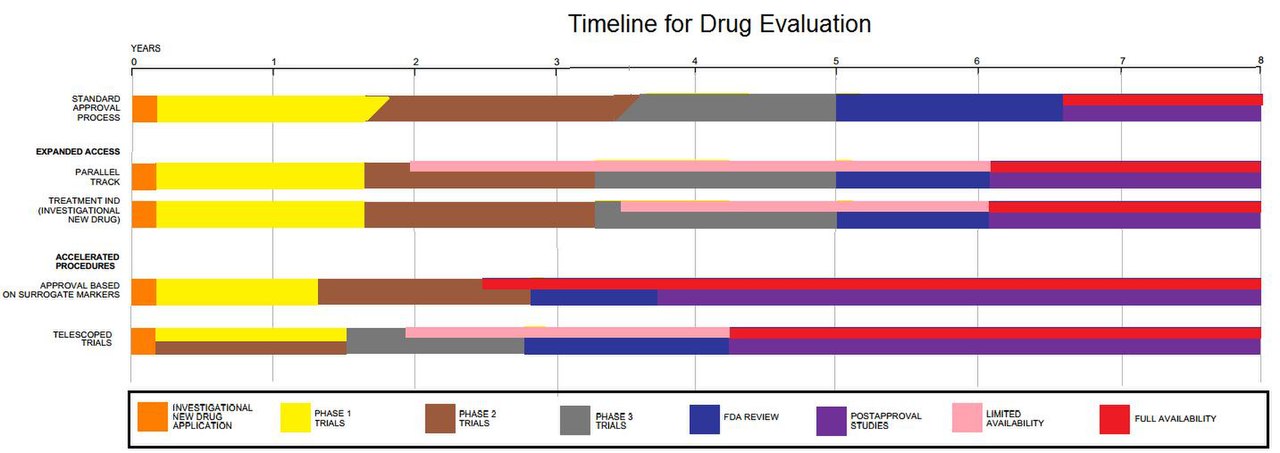
Cronograma para la evaluación de medicamentos
Las IND de investigación o de investigación son IND no comerciales presentadas por investigadores para estudiar un medicamento no aprobado o para estudiar un medicamento aprobado para una nueva indicación o en una nueva población de pacientes.
Las solicitudes de uso de emergencia , también llamadas solicitudes de uso compasivo o solicitudes de uso de un solo paciente, se presentan para el uso de emergencia de un medicamento no aprobado cuando la situación clínica no permite tiempo suficiente para presentar una solicitud de uso de emergencia de acuerdo con los artículos 312.23 y 312.24 del Título 21 del CFR. Se utilizan con mayor frecuencia para afecciones potencialmente mortales para las que no existe un tratamiento estándar.
Las solicitudes de autorización de comercialización de tratamiento se presentan para que un medicamento esté disponible para el tratamiento de afecciones graves o que pongan en riesgo la vida de inmediato antes de la aprobación de la FDA. Las enfermedades o afecciones graves son los accidentes cerebrovasculares, la esquizofrenia, la artritis reumatoide, la osteoartritis, la depresión crónica, las convulsiones, la demencia de Alzheimer, la esclerosis lateral amiotrófica (ELA) y la narcolepsia.
Las solicitudes de patente internacional de detección se presentan para varios compuestos estrechamente relacionados con el fin de detectar los compuestos o formulaciones preferidos. El compuesto preferido puede entonces desarrollarse bajo una solicitud de patente internacional independiente. Se utiliza para detectar diferentes sales, ésteres y otros derivados de fármacos que son químicamente diferentes, pero farmacodinámicamente similares.
Solicitud
La solicitud de IND se puede dividir en las siguientes categorías: [2]
Las pruebas preclínicas consisten en estudios farmacológicos y toxicológicos en animales para evaluar si el medicamento es seguro para su uso en humanos. También se incluye cualquier experiencia previa con el medicamento en humanos (a menudo, uso en el extranjero).
La información de fabricación incluye la composición, el fabricante y la estabilidad del medicamento, así como los controles utilizados para su fabricación. Se utiliza para garantizar que la empresa pueda producir y suministrar lotes adecuados y consistentes del medicamento.
Información del investigador sobre las cualificaciones de los investigadores clínicos, es decir, los profesionales (generalmente médicos) que supervisan la administración del fármaco experimental a los sujetos del estudio. Se utiliza para evaluar si los investigadores están cualificados para cumplir con sus funciones en el ensayo clínico.
Los protocolos de ensayos clínicos son la pieza central del IND. Protocolos detallados para los estudios clínicos propuestos para evaluar si los ensayos de la fase inicial expondrán a los sujetos a riesgos innecesarios.
Otros compromisos son el de obtener el consentimiento informado de los sujetos de investigación, el de obtener una revisión del estudio por parte de una junta de revisión institucional (IRB) y el de cumplir con las regulaciones sobre nuevos medicamentos en investigación.
Una solicitud de IND también debe incluir un Folleto para el investigador destinado a educar a los investigadores del ensayo sobre los hechos importantes sobre el medicamento del ensayo que necesitan saber para llevar a cabo su ensayo clínico con el menor riesgo para los sujetos o pacientes. [ cita requerida ]
Una vez que se presenta una solicitud de IND, la FDA tiene 30 días para objetar la IND o esta entrará en vigencia automáticamente y podrán comenzar los ensayos clínicos . Si la FDA detecta un problema, puede suspender la IND, prohibiendo el inicio de los estudios clínicos hasta que se resuelva el problema, como se describe en 21 CFR 312.42 .
Un IND debe tener la etiqueta “Precaución: nuevo fármaco: limitado por la ley federal (o de los Estados Unidos) a uso investigativo”, según 21 CFR 312.6
La FDA tiene un programa IND ( Compassionate Investigational New Drug Program ) para el uso medicinal de la marihuana . En 1992 dejó de aceptar nuevos pacientes, después de que las autoridades de salud pública concluyeran que no tenía valor científico y debido al deseo de la administración del presidente George H. W. Bush de "ponerse firme contra el crimen y las drogas". En 2011, cuatro pacientes seguían recibiendo cannabis del gobierno en virtud de este programa. [3]
^ "Consideraciones generales sobre la E8(R1) para estudios clínicos" (PDF) . Directrices de eficacia . ICH.
^ John S. McInnes (2011). "Nuevas aplicaciones de fármacos". En Shayne C. Gad (ed.). Nuevas aplicaciones de fármacos . Enciclopedia de ciencias farmacéuticas . doi :10.1002/9780470571224.pse420. ISBN9780470571224.
^ "4 estadounidenses obtienen marihuana medicinal de manos del gobierno federal". CBS News . 28 de septiembre de 2011.
Enlaces externos
Proceso de solicitud de nuevo fármaco en investigación (IND) Centro de Evaluación e Investigación de Medicamentos, Administración de Alimentos y Medicamentos.
Guía de la ICH para la industria, E6 Buenas prácticas clínicas: guía consolidada. ENLACE ROTO
Troetel, WM: Cómo lograr una presentación exitosa de un IND en EE. UU. (1) The Regulatory Affairs Journal. 6: 22–28, enero de 1995.
Troetel, WM: Cómo lograr una presentación exitosa de un IND en EE. UU. (2) The Regulatory Affairs Journal. 6: 104–108, febrero de 1995.