
La reacción de sustitución nucleofílica unimolecular ( S N 1 ) es una reacción de sustitución en química orgánica . El símbolo de Hughes-Ingold del mecanismo expresa dos propiedades: "S N " significa " sustitución nucleofílica " y el "1" dice que el paso que determina la velocidad es unimolecular . [1] [2] Por lo tanto, la ecuación de velocidad a menudo se muestra como que tiene una dependencia de primer orden del sustrato y una dependencia de orden cero del nucleófilo . Esta relación se mantiene para situaciones en las que la cantidad de nucleófilo es mucho mayor que la del intermedio. En cambio, la ecuación de velocidad se puede describir con mayor precisión utilizando cinética de estado estacionario . La reacción involucra un intermedio carbocatión y se observa comúnmente en reacciones de haluros de alquilo secundarios o terciarios en condiciones fuertemente básicas o, en condiciones fuertemente ácidas, con alcoholes secundarios o terciarios . Con haluros de alquilo primarios y secundarios, ocurre la reacción alternativa S N 2 . En química inorgánica , la reacción S N 1 se conoce a menudo como sustitución disociativa . Esta vía de disociación está bien descrita por el efecto cis . Un mecanismo de reacción fue introducido por primera vez por Christopher Ingold et al. en 1940. [3] Esta reacción no depende mucho de la fuerza del nucleófilo, a diferencia del mecanismo S N 2. Este tipo de mecanismo implica dos pasos. El primer paso es la ionización del haluro de alquilo en presencia de acetona acuosa o alcohol etílico. Este paso proporciona un carbocatión como intermediario.
En el primer paso del mecanismo S N 1, se forma un carbocatión que es plano y, por lo tanto, el ataque del nucleófilo (segundo paso) puede ocurrir desde cualquier lado para dar un producto racémico, pero en realidad no se produce una racemización completa. Esto se debe a que la especie nucleófila ataca al carbocatión incluso antes de que el ion haluro que se aleja se haya alejado lo suficiente del carbocatión. El ion haluro cargado negativamente protege al carbocatión de ser atacado en el lado frontal, y se prefiere el ataque en el lado posterior, que conduce a la inversión de la configuración. Por lo tanto, el producto real sin duda consiste en una mezcla de enantiómeros, pero predominarían los enantiómeros con configuración invertida y no se produce una racemización completa. [4]
Un ejemplo de una reacción que tiene lugar con un mecanismo de reacción S N 1 es la hidrólisis del bromuro de terc-butilo formando terc -butanol :

Esta reacción S N 1 tiene lugar en tres pasos:




Aunque la ley de velocidad de la reacción S N 1 suele considerarse de primer orden en el caso de los haluros de alquilo y de orden cero en el caso de los nucleófilos, se trata de una simplificación que solo es válida en determinadas condiciones. Si bien también es una aproximación, la ley de velocidad derivada de la aproximación de estado estacionario (SSA) proporciona más información sobre el comportamiento cinético de la reacción S N 1. Considere el siguiente esquema de reacción para el mecanismo que se muestra arriba:

Aunque es un carbocatión terciario relativamente estable , el catión terc -butilo es una especie de alta energía que está presente solo en concentraciones muy bajas y no se puede observar directamente en condiciones normales. Por lo tanto, el SSA se puede aplicar a esta especie:
(1) Supuesto de estado estacionario:
(2) Concentración del catión t-butilo, basada en el supuesto de estado estacionario:
(3) Velocidad de reacción global, suponiendo un paso final rápido:
(4) Ley de velocidad en estado estacionario, sustituyendo (2) por (3):
En condiciones sintéticas normales, el nucleófilo entrante es más nucleófilo que el grupo saliente y está presente en exceso. Además, los experimentos cinéticos se realizan a menudo en condiciones de velocidad inicial (conversión del 5 al 10 %) y sin la adición de bromuro, por lo que es insignificante. Por estas razones, a menudo se cumple. En estas condiciones, la ley de velocidad SSA se reduce a:
La sencilla ley de velocidad de primer orden descrita en los libros de texto introductorios. En estas condiciones, la concentración del nucleófilo no afecta a la velocidad de la reacción, y el cambio de nucleófilo (por ejemplo, de H2O a MeOH) no afecta a la velocidad de la reacción, aunque el producto es, por supuesto, diferente. En este régimen, el primer paso (ionización del bromuro de alquilo) es lento, determinante de la velocidad e irreversible, mientras que el segundo paso (adición nucleofílica) es rápido y cinéticamente invisible.
Sin embargo, en determinadas condiciones, se pueden observar reacciones cinéticas que no son de primer orden. En particular, cuando hay una gran concentración de bromuro y la concentración de agua es limitada, la reacción inversa del primer paso se vuelve importante cinéticamente. Como indica la ley de velocidad SSA, en estas condiciones hay una dependencia fraccionaria (entre el orden cero y el primer orden) de [H 2 O], mientras que hay una dependencia fraccionaria negativa de [Br – ]. Por lo tanto, a menudo se observa que las reacciones S N 1 se ralentizan cuando se añade una fuente exógena del grupo saliente (en este caso, bromuro) a la mezcla de reacción. Esto se conoce como el efecto del ion común y la observación de este efecto es evidencia de un mecanismo S N 1 (aunque la ausencia de un efecto del ion común no lo descarta). [6] [7]
El mecanismo S N 1 tiende a predominar cuando el átomo de carbono central está rodeado de grupos voluminosos, ya que dichos grupos obstaculizan estéricamente la reacción S N 2. Además, los sustituyentes voluminosos en el carbono central aumentan la velocidad de formación del carbocatión debido al alivio de la tensión estérica que se produce. El carbocatión resultante también se estabiliza mediante la estabilización inductiva y la hiperconjugación de los grupos alquilo unidos . El postulado de Hammond-Leffler sugiere que esto también aumentará la velocidad de formación del carbocatión. Por lo tanto, el mecanismo S N 1 predomina en las reacciones en los centros alquilo terciarios .
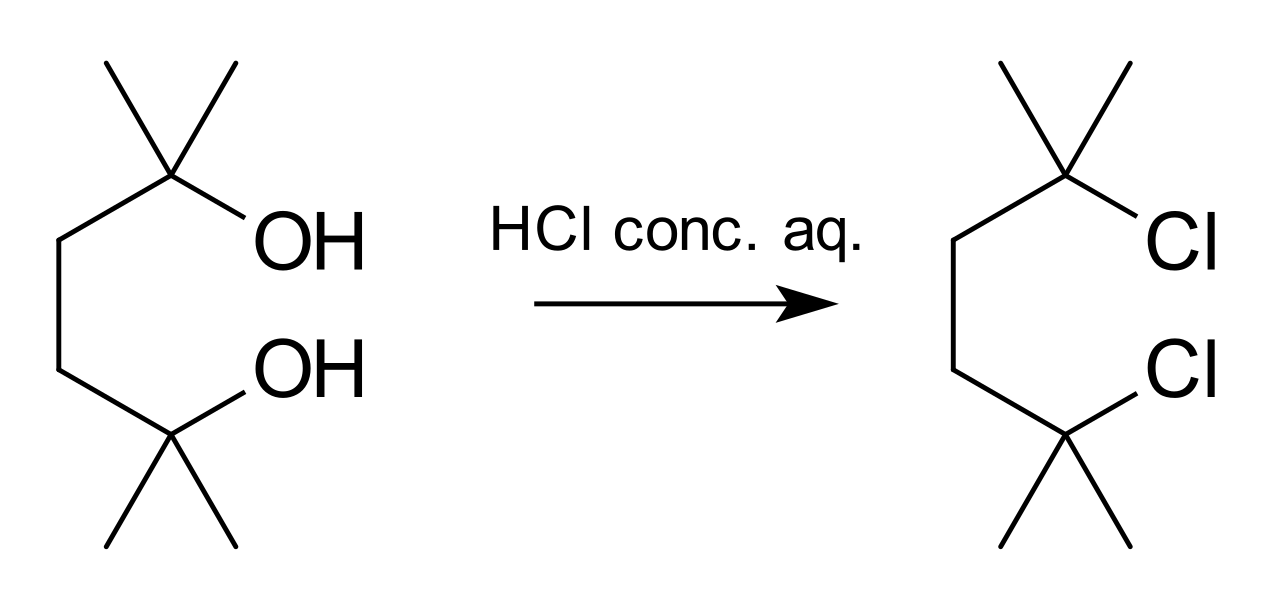
Un ejemplo de una reacción que se desarrolla de manera S N 1 es la síntesis de 2,5-dicloro-2,5-dimetilhexano a partir del diol correspondiente con ácido clorhídrico concentrado : [8]
A medida que aumentan las sustituciones alfa y beta con respecto a los grupos salientes, la reacción se desvía de S N 2 a S N 1.
El carbocatión intermedio formado en el paso de determinación de la velocidad de la reacción (RDS) es un carbono hibridado sp2 con geometría molecular plana trigonal. Esto permite dos formas diferentes de ataque nucleofílico, una en cada lado de la molécula plana. Si no se prefiere ninguno de los enfoques, entonces estas dos formas ocurren por igual, produciendo una mezcla racémica de enantiómeros si la reacción tiene lugar en un estereocentro. [9] Esto se ilustra a continuación en la reacción S N 1 de S-3-cloro-3-metilhexano con un ion yoduro, que produce una mezcla racémica de 3-yodo-3-metilhexano:

Sin embargo, se puede observar un exceso de un estereoisómero, ya que el grupo saliente puede permanecer cerca del carbocatión intermedio durante un corto tiempo y bloquear el ataque nucleofílico. Esto contrasta con el mecanismo S N 2 , que es un mecanismo estereoespecífico donde la estereoquímica siempre está invertida ya que el nucleófilo entra desde el lado posterior del grupo saliente.
Dos reacciones secundarias comunes son las reacciones de eliminación y la transposición de carbocatión . Si la reacción se realiza en condiciones cálidas o calientes (que favorecen un aumento de la entropía), es probable que predomine la eliminación de E1 , lo que lleva a la formación de un alqueno . A temperaturas más bajas, las reacciones S N 1 y E1 son reacciones competitivas y se vuelve difícil favorecer una sobre la otra. Incluso si la reacción se realiza en frío, se puede formar algún alqueno. Si se intenta realizar una reacción S N 1 utilizando un nucleófilo fuertemente básico como el ion hidróxido o metóxido , el alqueno se formará nuevamente, esta vez a través de una eliminación de E2 . Esto será especialmente cierto si la reacción se calienta. Finalmente, si el intermedio de carbocatión puede transponerse a un carbocatión más estable, dará un producto derivado del carbocatión más estable en lugar del producto de sustitución simple.
Dado que la reacción S N 1 implica la formación de un carbocatión intermedio inestable en el paso determinante de la velocidad (RDS), cualquier cosa que pueda facilitar este proceso acelerará la reacción. Los disolventes normales de elección son tanto polares (para estabilizar los intermedios iónicos en general) como disolventes próticos (para solvatar el grupo saliente en particular). Los disolventes próticos polares típicos incluyen agua y alcoholes, que también actuarán como nucleófilos, y el proceso se conoce como solvólisis.
La escala Y correlaciona las tasas de reacción de solvólisis de cualquier solvente ( k ) con la de un solvente estándar (80% v/v etanol / agua ) ( k 0 ) a través de
con m una constante de reactivo (m = 1 para cloruro de terc -butilo ) e Y un parámetro de solvente. [10] Por ejemplo, 100% de etanol da Y = −2,3, 50% de etanol en agua Y = +1,65 y 15% de concentración Y = +3,2. [11]
{{cite book}}: CS1 maint: nombres múltiples: lista de autores ( enlace ) CS1 maint: nombres numéricos: lista de autores ( enlace )![{\displaystyle {\frac {d[{\text{tBu}}^{+}]}{dt}}=0=k_{1}[{\text{tBuBr}}]-k_{-1}[{\text{tBu}}^{+}][{\text{Br}}^{-}]-k_{2}[{\text{tBu}}^{+}][{\text{H}}_{2}{\text{O}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a3c62114166c94c369041396194a6d43191c6c20)
![{\displaystyle [{\text{tBu}}^{+}]={\frac {k_{1}[{\text{tBuBr}}]}{k_{-1}[{\text{Br}}^{-}]+k_{2}[{\text{H}}_{2}{\text{O}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/61023e015605bc9434b1acd07bd467d73baecb57)
![{\displaystyle {\frac {d[{\text{tBuOH}}]}{dt}}=k_{2}[{\text{tBu}}^{+}][{\text{H}}_{2}{\text{O}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/01d5344985204038591421721477b492b4e4aed6)
![{\displaystyle {\frac {d[{\text{tBuOH}}]}{dt}}={\frac {k_{1}k_{2}[{\text{tBuBr}}][{\text{H}}_{2}{\text{O}}]}{k_{-1}[{\text{Br}}^{-}]+k_{2}[{\text{H}}_{2}{\text{O}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e220b039163872dd2472550959f71920b622092b)
![{\displaystyle [{\text{Br}}^{-}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/28d340f11608b070d0e0968bdc2c0c090e939937)
![{\displaystyle k_{-1}[{\text{Br}}^{-}]\ll k_{2}[{\text{H}}_{2}{\text{O}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c264ca5dd227854e9894f4b9ff597898d75bd752)
![{\displaystyle {\text{tasa}}={\frac {d[{\text{tBuOH}}]}{dt}}={\frac {k_{1}k_{2}[{\text{tBuBr}}][{\text{H}}_{2}{\text{O}}]}{k_{2}[{\text{H}}_{2}{\text{O}}]}}=k_{1}[{\text{tBuBr}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/76290a9c4e1b075244789b3e8943564103af557c)
