La regulación epigenética de la neurogénesis es el papel que juega la epigenética (características hereditarias que no implican cambios en la secuencia de ADN) en la regulación de la neurogénesis (la producción de neuronas a partir de células madre neurales ).
La epigenética es el estudio de los cambios hereditarios en la expresión genética que no resultan de modificaciones en la secuencia del ADN . La neurogénesis es el mecanismo de proliferación y diferenciación neuronal . Implica muchos procesos complejos diferentes que dependen del tiempo y del orden. [1]
Procesos como la proliferación neuronal, la especificación del destino, la diferenciación, la maduración y la integración funcional de las células recién nacidas en las redes neuronales existentes están todos interconectados. [2] En la última década [ ¿cuándo? ] se ha demostrado que muchos mecanismos reguladores epigenéticos desempeñan un papel importante en la cronología y la determinación de los linajes de células madre neuronales . [1]

Tres métodos importantes de regulación epigenética incluyen la modificación de histonas , la metilación y desmetilación del ADN y la expresión de microARN ( miARN ). Las histonas mantienen el ADN de la célula eucariota firmemente empaquetado a través de interacciones de carga entre la carga positiva en la cola de la histona y la carga negativa del ADN, así como entre las colas de histonas de los nucleosomas cercanos . Si bien hay muchos tipos diferentes de modificaciones de histonas, en la epigenética neuronal hay dos mecanismos principales que se han explorado: la metilación de histonas y la acetilación de histonas . [1] [3] En la primera, se agregan o eliminan grupos metilo a la histona alterando su estructura y exponiendo la cromatina y llevando a la activación o desactivación del gen. En el segundo, la acetilación de histonas hace que la histona sostenga el ADN de manera más suelta, lo que permite una mayor activación del gen. La metilación del ADN, en la que se añaden grupos metilo a los residuos de citosina o adenosina en el ADN, es un método más duradero de inactivación genética que la modificación de histonas, aunque sigue siendo reversible en algunos casos. [1] [3] Los microARN son una forma pequeña de ARN no codificante (ARNnc) que a menudo actúan como mecanismos de "ajuste fino" para la expresión genética al reprimir o inducir el ARN mensajero (ARNm) en las células neuronales, pero también pueden actuar directamente con factores de transcripción para guiar la neurogénesis. [1] [3] [4] [5]
Las células madre neuronales participan en el desarrollo de la corteza de una manera precisa "de adentro hacia afuera" con mecanismos de sincronización cuidadosamente controlados. Las neuronas que nacen temprano forman capas profundas en la corteza, mientras que las neuronas que nacen más recientemente forman las capas superiores. Este programa de sincronización se observa tanto in vitro como in vivo. [1] [3] El análisis de mutantes ha demostrado que la metilación de histonas modula la producción de neuronas de la capa profunda y de la capa superior a través de la regulación epigenética. Específicamente, la eliminación de una parte del complejo PRC2 , Ezh2 , que codifica la metiltransferasa de histonas , condujo a una reducción al doble de las neuronas de la capa superior que expresaban POU3F2 /BRN2 y que expresaban SATB2 sin afectar el número de neuronas en las capas V y VI. En los progenitores neuronales derivados de células madre embrionarias de ratón, el aumento de la acetilación de histonas inducida por el inhibidor de la histona desacetilasa (HDAC), el ácido valproico , no solo indujo la diferenciación neuronal, sino que también enriqueció selectivamente la población neuronal de la capa superior. Por lo tanto, se ha propuesto que la inhibición de la HDAC promueve la progresión de la diferenciación neuronal, lo que conduce a un cambio de destino de los progenitores productores de la capa profunda a los progenitores de la capa superior. Sin embargo, las razones detrás de esta diferenciación selectiva y el control del tiempo como resultado de la inhibición de la HDAC aún no se comprenden por completo. [3]
La naturaleza crítica de la metilación del ADN para la corticogénesis se ha demostrado a través de experimentos de knock out en ratones. Cuando DNMT 3b y DNMT1 se eliminaron por separado en embriones de ratón, murieron debido al deterioro del desarrollo del tubo neural . El silenciamiento de DNMT3a no causó letalidad embrionaria, pero resultó en un detrimento severo en la neurogénesis posnatal. [1] [3] Esto se debe en gran medida al momento en que estos mecanismos epigenéticos están activos. DNMT3b se expresa en células progenitoras neuronales tempranas y disminuye a medida que avanza el desarrollo neuronal y DNMT3a es apenas detectable hasta el día embrionario 10 (E.10). Sin embargo, en E.10, la expresión de DNMT3a aumenta significativamente desde E13.5 y hasta bien entrada la edad adulta. En el prosencéfalo posnatal, DNMT3a se expresa en la zona subventricular (SVZ) y el giro dentado del hipocampo , las ubicaciones primarias para la neurogénesis adulta. [1] [2] La pérdida de DNMT3a en las células progenitoras neuronales postnatales conduce a la regulación negativa de genes neurónicos como Dlx2 , Neurog2 y Sp8 ; pero a la regulación positiva de genes involucrados en la diferenciación astroglial y oligodendroglial , lo que indica un papel en el cambio de destino celular de neurogénesis a gliogénesis . La desmetilación del ADN, así como la metilación de ciertos genes, permite que la neurogénesis proceda de manera dependiente del tiempo. Uno de estos genes es Hes5 , hipermetilado en embriones E7.5 pero completamente desmetilado por E9.5, que es uno de los genes diana en la vía de señalización Notch . GCM1 y GCM2 desmetilan el promotor Hes5 , lo que le permite responder a la señalización NOTCH e iniciar la generación de células madre neuronales. [1] Otro ejemplo es el gen Gfap que es necesario para la diferenciación de los astrocitos. La capacidad de diferenciarse en células gliales está reprimida en las células madre neuronales con un destino celular neuronal. Esta represión se debe en gran medida a la falta de respuesta de las células madre neuronales a los estímulos que inducen la formación de astrocitos. Las células madre neuronales no responden debido al ADN hipermetilado en las regiones promotoras de los genes de los astrocitos, como Gfap . El sitio de unión de STAT3 en la región promotora de Gfap está hipermetilado en E11.5 y apenas en E14.5, momento en el que puede recibir estímulos que inducen la formación de astrocitos y comenzar la diferenciación de astrocitos inducible por citocinas. [3]
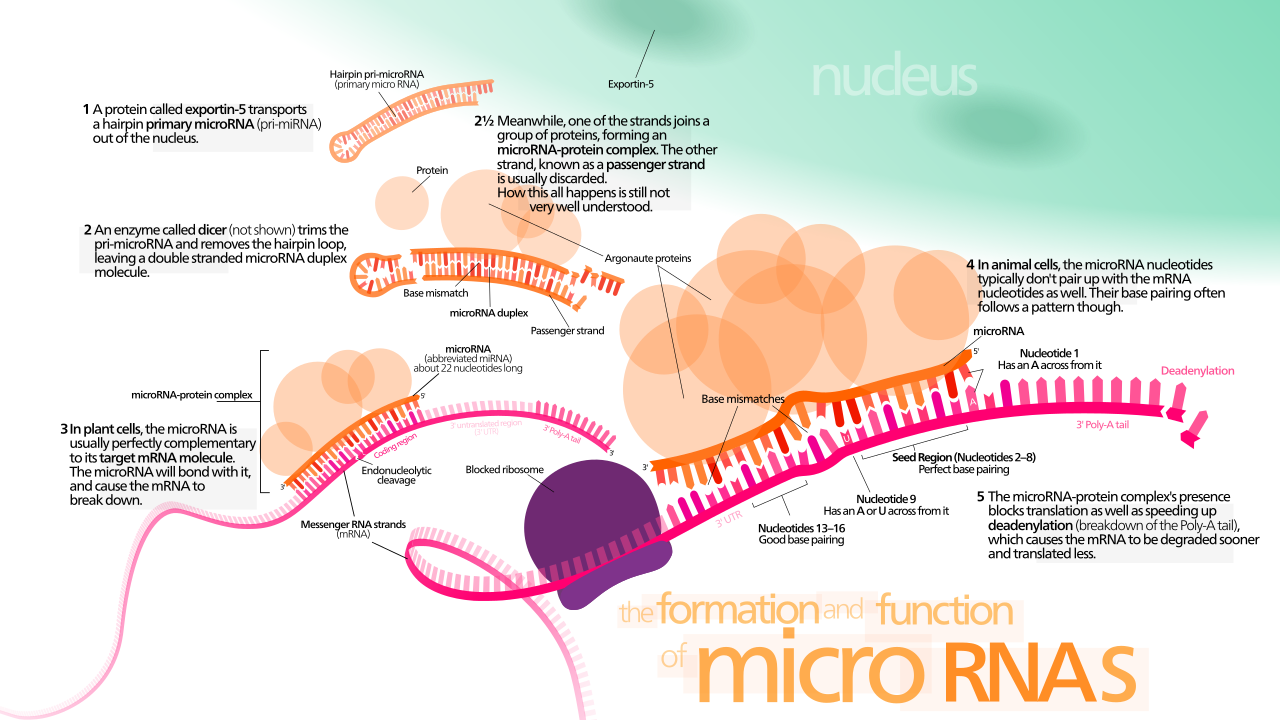
Estudios realizados por De Pietri Tonelli y Kawase-Koga han demostrado que la inactivación condicional de Dicer , una enzima ampliamente utilizada para la síntesis de miRNA, en el neocórtex de ratón resultó en un tamaño cortical reducido, mayor apoptosis neuronal y una deficiente estratificación de cortisol. Las células neuroepiteliales y las células neuroprogenitoras no se vieron afectadas hasta E.14, momento en el que también experimentaron apoptosis. Esto no muestra qué miRNA fueron responsables de los diversos factores afectados, pero sí muestra que existe un requisito específico de la etapa para la expresión de miRNA en el desarrollo cortical. [1] [5] [6] [7] miR-124 , el microRNA más abundante en el sistema nervioso central, controla la progresión del linaje de las células progenitoras neuronales de la zona subventricular en neuroblastos al suprimir la producción de proteínas al dirigirse a Sox9 . Otro actor importante de microRNA es miR-9/9*. En la neurogénesis embrionaria, se ha demostrado que miR-9 regula la diferenciación neuronal y la autorrenovación. [1] [4] [5] La expresión ectópica de miR-9 en la corteza del ratón en desarrollo provocó una diferenciación neuronal prematura e interrumpió la migración de nuevas neuronas a través de la focalización de Foxg1 . [1]
Contrariamente a la idea de que los microARN son sólo mecanismos de ajuste fino, estudios recientes han demostrado que miR-9 y miR-124 pueden actuar juntos para guiar a los fibroblastos hacia las células neuronales. Los factores de transcripción y los genes reguladores, como Neurod1 , Ascr1 y Myt1l , que anteriormente se creía que eran responsables de este fenómeno, no transformaron fibroblastos humanos en ausencia de miR-9 y miR-124, pero en presencia de los microARN y la ausencia de los factores de transcripción, la transformación de fibroblastos humanos se produjo, aunque de una manera menos eficiente. [1] [4] [5]
La neurogénesis continúa después del desarrollo hasta bien entrada la edad adulta. [2] La beta (GADD45b), inducible por daño del ADN y detención del crecimiento, es necesaria para la desmetilación de los promotores de genes críticos responsables del desarrollo de neuronas recién nacidas, como el factor neurotrófico derivado del cerebro (BDNF) y el factor de crecimiento básico de fibroblastos (FGF2). [8] Como tal, la regulación positiva de GADD45b conduce a un aumento de la desmetilación, un aumento del BDNF y del FGF2 y, en última instancia, a un mayor número de células progenitoras neuronales. [1] [8]
La acetilación y desacetilación de histonas , así como la inhibición de los mecanismos de desacetilación de histonas, también desempeñan papeles importantes en la proliferación y autorrenovación de las células madre neuronales postnatales. El ADN , que codifica genes dentro del genoma, incluidos los involucrados en la neurogénesis adulta, se empaqueta en la cromatina . La cromatina en sí está formada por subunidades de nucleosomas , cada una de las cuales consta de dos copias de cada una de las proteínas histonas H2A , H2B , H3 y H4 . Una de las funciones principales que desempeña la acetilación en la regulación de la expresión génica es a través de la inhibición de las interacciones de nucleosomas adyacentes. Cuando las histonas H4 no están acetiladas, son de naturaleza básica y se insertan en el bolsillo ácido de los dímeros de proteína H2A-H2B en nucleosomas adyacentes, lo que lleva a una asociación estrecha entre nucleosomas y un mayor empaquetamiento de la cromatina. Por lo tanto, la acetilación hace que la histona H4 pierda su basicidad y evita la reticulación de nucleosomas. [9] Esta acetilación de las colas de histonas aumenta adicionalmente la afinidad de los complejos enzimáticos de remodelación de la cromatina, como SWI-SNF e ISWI, que utilizan ATP para producir regiones libres de nucleosomas en los sitios promotores y potenciadores. [10] Esto permite una mayor capacidad de reconocimiento de estos sitios por los factores de transcripción, particularmente TFIID , que es el principal factor de transcripción involucrado en la iniciación de la transcripción . Además, la acetilación de los residuos de lisina en las colas de histonas puede ser reconocida por el componente TAF1 de TFIID, y cuando se une, TAF1 se convierte en una acetiltransferasa de histonas (HAT), acetilando aún más las histonas H3 y H4 adyacentes y reclutando más HAT en el proceso. [11] El ADN envuelve las histonas de la cromatina, y la acetilación de estas colas de histonas conduce a la reducción de la carga positiva asociada con las histonas. Esto hace que el ADN cargado negativamente pierda afinidad por la histona, lo que permite que haya más espacio para que los factores de transcripción se unan a las regiones promotoras y faciliten aún más la expresión.
Finalmente, los procesos de acetilación de histonas y la consiguiente remodelación de la cromatina permiten una mayor expresión de genes diana, incluidos los implicados en la neurogénesis adulta. Los reguladores de la remodelación de la cromatina más estudiados y mejor comprendidos, que desempeñan un papel importante en la neurogénesis adulta, son las histonas acetiltransferasas (HAT) y las histonas desacetilasas (HDAC). Las HAT añaden grupos acetilo a los nucleosomas, mientras que las HDAC los eliminan. La acetilación de las histonas conduce a una menor condensación de los nucleosomas al ADN diana y aumenta la probabilidad de que se produzca la expresión génica al liberar los objetivos de ADN para que se unan a sus respectivos factores de transcripción. Este proceso está involucrado en la regulación de la proliferación neuronal, ya que se expresan y reprimen diferentes genes de las células neuronales. La desacetilación de las histonas conduce a lo contrario y aumenta la probabilidad de represión de la expresión génica. [12]
Los inhibidores de HDAC (HDACi), como el ácido valproico (VPA) y la tricostatina A, pueden promover la proliferación de la neurogénesis adulta a través de la inhibición de la actividad de HDAC, induciendo la diferenciación de células progenitoras adultas. [12] Las HDAC expresadas en los nervios interactúan con Tlx, un regulador esencial de las células madre neuronales, para suprimir los genes diana de TLX. Esto incluye el inhibidor de la cinasa dependiente de ciclina P21 y el gen supresor de tumores Pten para promover la proliferación de células madre neuronales. [1] La inhibición de las HDAC por el fármaco antiepiléptico ácido valproico induce la diferenciación neuronal como en la neurogénesis embrionaria, pero también inhibe la diferenciación de células gliales de las células madre neuronales adultas. Esto probablemente esté mediado por la regulación positiva de genes neuronales específicos como los factores de transcripción neurogénicos básicos hélice-bucle-hélice NEUROD, NEUROGENENIN1 y Math1. La pérdida condicional de HDAC1 y HDAC2 en las células progenitoras neuronales impidió que se diferenciaran en neuronas y su pérdida en las células progenitoras oligodendríticas interrumpió la formación de oligodendrocitos, lo que sugiere que la desacetilación de histonas desempeña funciones importantes pero variables en diferentes etapas del desarrollo neuronal. [1]
Los microARN ( miARN ) son pequeños ARN no codificantes que desempeñan un papel importante en la regulación epigenética eucariota. Los miARN funcionan para modular los niveles de expresión de proteínas de sus dianas de ARNm sin afectar las secuencias de los genes de interés. Si bien los miARN desempeñan un papel importante en la modulación de los mecanismos epigenéticos, también son modificados y regulados por otros factores epigenéticos, incluida la metilación del ADN, las modificaciones de las histonas y otras modificaciones del ARN . [13] Juntos, los miARN crean un ciclo de retroalimentación epigenética con otros factores epigenéticos para afectar los niveles de expresión de genes específicos. Se ha implicado a varios miARN específicos como agentes de regulación epigenética en la neurogénesis adulta. miR-9 se dirige al receptor nuclear TLX en la neurogénesis adulta para promover la diferenciación neuronal e inhibir la proliferación de células madre neuronales. También influye en la especificación del subtipo neuronal y regula el crecimiento axonal, la ramificación y la focalización en el sistema nervioso central a través de interacciones con HES1 , una molécula de homeostasis de células madre neuronales. El miR-124 promueve la salida del ciclo celular y la diferenciación neuronal en la neurogénesis adulta. Estudios en ratones han demostrado que la expresión ectópica del miR-124 provocó una diferenciación prematura de las células progenitoras neuronales y un agotamiento en la zona subventricular .
Además de miR-9 y miR-124, otros miRNAs desempeñan papeles esenciales en la regulación de la neurogénesis adulta. miR-137, miR-184 y miR-195 regulan la proliferación de células madre neurales adultas, con su sobreexpresión conduciendo a una proliferación regulada al alza mientras que su regulación a la baja conduce a una disminución en la proliferación neuronal. [14] La proteína de unión a metil-CpG 1 ( MBD1 ) reprime miR-184, que es un microRNA responsable de la proliferación de células madre/progenitoras neurales adultas (aNSCs) junto con la inhibición de la diferenciación de estas células. miR-184 regula el desarrollo cerebral embrionario uniéndose al ARNm para la proteína Numblike (Numbl) y alterando su expresión. MBD1, Numbl y miR-184 trabajan todos juntos para regular la proliferación y diferenciación de aNSC. [15] Además, el miR-195 trabaja en estrecha colaboración con el MBD1 para regular la proliferación y diferenciación de las células madre neurales autólogas. El mIR-194 y el MBD1 forman un circuito regulador negativo en las células madre neurales autólogas y trabajan para reprimir la expresión de cada uno de ellos. La inhibición del miR-195 promueve la diferenciación de las células madre neurales autólogas. Una vez que se ha producido la diferenciación, los niveles de miR-195 disminuyen. [16]
Los astrocitos son células gliales que forman la barrera hematoencefálica, sostienen las sinapsis y guían axones. [17] A diferencia de las neuronas, estas células gliales especializadas pueden alterar su destino celular antes de alcanzar la maduración completa y " desdiferenciarse ", en gran parte debido a factores epigenéticos. Esta desdiferenciación permite que los astrocitos alcancen potencialmente un destino celular completamente diferente, siempre que esta desdiferenciación ocurra antes de que ocurra la maduración completa, y puede conducir a su consecuente diferenciación y conversión de células gliales en neuronas en el cerebro adulto. [18] Antes de la maduración completa, mientras la desdiferenciación aún es posible, la expresión de los genes Mash1, NeuroG1 y NeuroG2 puede reprogramar los astrocitos en neuronas. [19] Además de la expresión de estos genes en las células cerebrales, múltiples factores epigenéticos juegan un papel en los patrones de expresión de estos genes. Se ha demostrado que una regulación positiva de la acetilación en los residuos H3K9 y H3K14 adyacentes a los genes NeuroG1 y NeuroG2 acompaña a la desdiferenciación de los astrocitos; la sobreexpresión y/o la expresión forzada de estos genes pueden inducir directamente la diferenciación de los astrocitos.
Además, el silenciamiento de los mecanismos de metilación, específicamente el silenciamiento de muchas clases de metiltransferasas de ADN, que están involucradas en el silenciamiento de la expresión, inhibe que las células progenitoras de los astrocitos se diferencien de nuevo a su destino original como células gliales. [20] A pesar de este conocimiento del mecanismo de represión de la metilación, la identidad de estos genes silenciados aún no se conoce por completo. Si bien esta represión general de la metilación es necesaria para prevenir la expresión de genes específicos necesarios para permitir que un astrocito madure por completo y alcance un destino de célula astrocítica, se encontró que la sobreexpresión de una metiltransferasa de histona específica, Ezh2, que cataliza la trimetilación de H3K27, reprime los genes necesarios para el mantenimiento de los astrocitos, lo que permite que la célula conserve su morfología de célula madre neural. Esto demuestra que la metilación diferencial por distintas metiltransferasas y su consiguiente represión o sobreexpresión tienen diferentes papeles en la desdiferenciación de los astrocitos para formar neuronas. Además, si bien no es suficiente para inducir la desdiferenciación de los astrocitos por sí sola, Ezh2 es necesaria para que los astrocitos se desdiferencian, ya que se les impide alcanzar la madurez completa en su destino celular original. Una vez en esta etapa inhibida, se ha demostrado que la expresión del gen NeuroD4 en estas células gliales específicas conduce a la formación neuronal, y por lo tanto a la neurogénesis, a partir de los astrocitos desdiferenciados en cerebros de mamíferos adultos. [18]
La familia de genes inducibles por daño del ADN y detención del crecimiento 45 (Gadd45) desempeña un papel importante en el hipocampo. Gadd45 facilita la potenciación a largo plazo del hipocampo y mejora la memoria persistente para el rendimiento motor, el condicionamiento aversivo y la navegación espacial. [21] Además, se ha demostrado que la metilación del ADN es importante para la modulación dependiente de la actividad de la neurogénesis adulta en el hipocampo, que está mediada por GADD45b. GADD45b parece actuar como un sensor en las neuronas maduras para los cambios ambientales que se expresan a través de estos cambios de metilación. [1] Esto se determinó examinando los efectos de la aplicación de un estímulo eléctrico al giro dentado (DG) del hipocampo en ratones normales y deficientes en GADD45b. En ratones normales, la aplicación de estimulación eléctrica al DG aumentó la neurogénesis al aumentar el BDNF. Sin embargo, en ratones deficientes en GADD45b, el estímulo eléctrico tuvo un efecto menor. Un examen más detallado reveló que alrededor del 1,4 % de las islas CpG en las neuronas DG se metilan y desmetilan activamente tras una descarga eléctrica. Esto demuestra que los estados de metilación posmitóticos de las neuronas no son estáticos y, dado que se ha demostrado que los equipos de descarga eléctrica como el utilizado en el estudio tienen efectos terapéuticos en pacientes humanos con depresión y otros trastornos psiquiátricos, sigue existiendo la posibilidad de que los mecanismos epigenéticos puedan desempeñar un papel importante en la fisiopatología de los trastornos neuropsiquiátricos. [2] [8] Tanto DNMT1 como DNMT3a son necesarios en conjunto para el aprendizaje, la memoria y la plasticidad sináptica. [8]
La desregulación epigenética, o alteraciones en la maquinaria epigenómica, puede causar que los procesos de metilación del ADN y acetilación de histonas se descontrolen. La maquinaria epigenética influye en la regulación de la diferenciación neuronal (es decir, la neurogénesis) [22] y también está involucrada en procesos relacionados con la consolidación de la memoria y el aprendizaje en individuos sanos. [23] El aumento de la edad puede producir varios cambios epigenéticos, como la heterocromatina global reducida, la remodelación de nucleosomas, las marcas de histonas alteradas y los cambios en la metilación del ADN. Por ejemplo, la pérdida de nucleosomas ocurre debido al envejecimiento porque las proteínas histonas centrales se pierden y se produce una menor síntesis de proteínas. [24] Como el envejecimiento es el principal riesgo para muchos trastornos neurológicos, la desregulación epigenética puede a su vez conducir a alteraciones en el nivel transcripcional de genes involucrados en la patogénesis de enfermedades degenerativas neuronales como la enfermedad de Parkinson , la enfermedad de Alzheimer , la enfermedad de Huntington , la esquizofrenia y la enfermedad bipolar . [1] [25]
La expresión de microARN es fundamental para la neurogénesis. En pacientes con enfermedad de Alzheimer, miR-9 y miR-128 están regulados positivamente, mientras que miR-15a está regulado negativamente. [4] Los pacientes con Alzheimer también muestran disminuciones en el factor neurotrófico derivado del cerebro, que se ha demostrado que se reprime a través de la metilación del ADN. [8] Aunque lo que se ha argumentado como la mayor evidencia de la influencia epigenética en el Alzheimer es el gen que controla la proteína responsable de la formación de la placa amiloide , App . Este gen tiene un contenido muy alto de GC en su región promotora , lo que significa que es muy susceptible a la metilación del ADN. Se ha demostrado que este sitio promotor reduce naturalmente la metilación con el envejecimiento, lo que ejemplifica los paralelismos entre el envejecimiento y el Alzheimer ya bien conocidos. [26] [27] Los metales pesados también parecen interferir con los mecanismos epigenéticos. Específicamente en el caso de la APP, se ha demostrado que la exposición al plomo en etapas tempranas de la vida causa una marcada sobreexpresión de la proteína APP, lo que lleva a una mayor cantidad de placa amiloide más adelante en la vida en el cerebro envejecido. [27]
La relación entre la metilación del ADN y la edad se ha investigado más a fondo en las regiones promotoras de varios genes relacionados con el Alzheimer en los cerebros de pacientes post mortem con enfermedad de Alzheimer de inicio tardío. Los pacientes mayores parecen tener una maquinaria epigenética más anormal que los pacientes más jóvenes, a pesar del hecho de que ambos habían muerto de Alzheimer. Aunque esto en sí mismo no es una prueba concluyente de nada, ha llevado a una teoría de deriva epigenética relacionada con la edad según la cual las anomalías en la maquinaria epigenética y la exposición a ciertos factores ambientales que ocurren en etapas más tempranas de la vida conducen a patrones aberrantes de metilación del ADN mucho más tarde, lo que contribuye a la predisposición esporádica a la enfermedad de Alzheimer. [27]
Las modificaciones de histonas también pueden tener un impacto en la enfermedad de Alzheimer, pero las diferencias entre los efectos de HDAC en cerebros de roedores en comparación con cerebros humanos tienen a los investigadores desconcertados. [27] A medida que el enfoque de las enfermedades neurodegenerativas comienza a cambiar hacia la farmacología epigenética, se puede esperar que las interacciones de las modificaciones de histonas con respecto a la neurogénesis se vuelvan más claras.
La acetilación de histonas ha recibido un apoyo cada vez mayor a lo largo de los años como un mecanismo propuesto a través del cual la desregulación epigenética conduce a cambios en la expresión genética que contribuyen a la EH. [28] Los estudios que analizan ratones con EH frente al tipo salvaje (WT) han demostrado que los loci genéticos específicos (Drd2, Penk1, Actb y Grin1) disminuyen los niveles de acetilación de histonas, lo que sugiere que una mutación del gen Huntington (HTT) y su sobreexpresión pueden ser la causa de esta desregulación epigenética.
Se ha pensado que los inhibidores de HDAC (HDACi) podrían revertir parcialmente los bajos niveles de acetilación observados en pacientes con HD. Se han realizado estudios preclínicos utilizando varios HDACi [como el ácido hidroxámico suberoxilanilida (SAHA), la tricostatina A (TSA), el fenilbutirato y el butirato de sodio (NaB)] que se dirigen a HDACI y HDACII. Aunque estos inhibidores mejoran algunos fenotipos de HD en ratones, como la neuropatología y la función motora , estos efectos beneficiosos no conducen a una conclusión sobre la necesidad definitiva de aumentar los niveles de acetilación en pacientes con HD. Sin embargo, la inactivación de un objetivo de SAHA, Hdac 4, alivia las complicaciones neurodegenerativas en ratones con HD a través de un mecanismo independiente de la transcripción que actúa sobre los procesos de agregación de Htt mutante, lo que puede indicar que existe un mecanismo que involucra proteínas no histonas. [29] El mecanismo propuesto a través del cual se especula que actúa SAHA es a través de un modelo de degradación del proteasoma mediado por RANBP2 , que probablemente surge como un resultado general de las acciones de los inhibidores de HDAC. En este mecanismo, se muestra que SAHA regula a la baja Hdac 4 a través de un aumento en la sumoilación , que luego es seguido por la activación de la degradación a través de una vía proteasomal. Este mecanismo revela la conectividad entre los procesos de acetilación, desacetilación y sumoilación. [30]
Hasta 2014, no se ha demostrado que el tratamiento con HDACi restablezca la expresión normal de los genes de identidad neuronal. [31] Sin embargo, actualmente se están realizando estudios clínicos que utilizan HDACi y los resultados están pendientes; los estudios de fase II muestran resultados prometedores para el uso seguro y tolerable de varios compuestos como el fenilbutirato.
También se han documentado efectos beneficiosos de HDACi no mediados por histonas en modelos de enfermedad de Parkinson , lo que sugiere mecanismos comunes entre varias enfermedades neurodegenerativas.
El análisis de la metilación del ADN mostró que existe una desregulación significativa de la metilación en las islas CpG en pacientes con EP en comparación con individuos sanos. Aunque esto se produjo en todo el genoma, también se produjo en muchos genes de riesgo de EP. [32]
También se ha demostrado que la metilación de la citosina en el ADN mitocondrial fluctúa con el tiempo debido a la variación de la edad, ya que hay un creciente cuerpo de literatura que vincula la metilación del ADNmt con el envejecimiento y el estrés oxidativo. [33] Un estudio de 2015 de Hashizume et al. mostró que los niveles de ARNm de SHMT2 se reducen significativamente en los fibroblastos de personas mayores en comparación con individuos más jóvenes. El estudio también indicó además que la disminución de los niveles de expresión génica de GCAT y SHMT2 a través de shRNA y siRNA , respectivamente, en los fibroblastos de pacientes jóvenes condujo a una disfunción de la cadena respiratoria típica de los individuos seniles, lo que sugiere que un mecanismo epigenético puede ser la causa del cambio fenotípico. Como las mitocondrias juegan un papel en el desarrollo de la EP, [34] una mayor investigación en el área ayudará a descubrir cualquier implicación que la metilación del ADN mitocondrial tenga en la patogénesis de la EP.
El uso de neuronas dopaminérgicas que se han aislado de los pacientes con EP indicó que hubo aumentos en la acetilación (en H2A, H3 y H4) en comparación con el grupo de control de edad. [32] Otro estudio que involucró células tratadas con MPP+ (un compuesto que puede causar un estado de enfermedad similar a los mamíferos y humanos con EP [35] ) y cerebros de ratones tratados con (MPP+) mostró niveles disminuidos de HDAC, así como en muestras de mesencéfalo de pacientes con EP. Esto se ve potencialmente debido a cómo MPP+ promueve la descomposición de HDAC1 y HDAC2 a través de la autofagia , un proceso corporal de reciclaje de células viejas para hacer lugar para células más nuevas y saludables. [36] Estos resultados apuntan hacia el estrés de las modificaciones de histonas con respecto a la remodelación de la cromatina y su implicación en la patogénesis de la EP.
Los miRNA también están surgiendo como contribuyentes relevantes a la neurodegeneración en la EP. En particular, los pacientes con EP de la corteza frontal han mostrado niveles más altos de LRRK2 y niveles más bajos de miR-205 en comparación con los individuos sanos. Al conectar esto con los hallazgos de la capacidad de miR-205 de unirse al UTR 3' del ARNm de LRRK2 y suprimir la expresión, así como la prevención de defectos por miR-205 después de la introducción de una mutación R1441G LRRK2, estos resultados apuntan hacia miR-205 y su papel regulador en la expresión de LRRK2, lo que a su vez sugiere un papel regulador en la patogénesis de la EP.
En otro estudio en el que el aumento de la acetilación de microtúbulos mediante inhibidores de la desacetilasa o la tubulina acetilasa αTAT1 mostró prevención de la asociación del mutante LRRK2 con microtúbulos, la inhibición de las desacetilasas HDAC6 y Sirt2 a través de procesos de eliminación rescató tanto el transporte axonal como el comportamiento locomotor. [37] Esto se conecta además con los mecanismos comunes que involucran a HDACi en varias enfermedades neurodegenerativas.
Los trastornos bipolares son muy complejos y hereditarios, lo que los convierte en un trastorno interesante para examinar en busca de modificaciones epigenéticas. La metilación del ADN , la hidroximetilación del ADN y las modificaciones de las histonas pueden contribuir a la formación del trastorno bipolar.
Por ejemplo, los estudios de gemelos monocigóticos revelaron que los individuos con trastorno bipolar tenían una menor metilación del gen similar a la peptidilprolil isomerasa E (PPIEL), lo que puede atribuirse a la transmisión de dopamina. Los estudios indicaron que la hipermetilación de SLC6A4, un gen transportador de serotonina, también está relacionada con el trastorno bipolar. Una mayor expresión de la ADN metiltransferasa 1 en las interneuronas GABAérgicas corticales puede permitir la hipermetilación. La hipermetilación puede provocar que se produzca hidroximetilación para compensar en exceso los efectos represivos de la hipermetilación. La metilación de las regiones CpG es relevante para los trastornos bipolares. Los pacientes con trastorno bipolar mostraron niveles más bajos de metilación para la región CpG del gen KCNQ3, que es responsable del canal de K+ dependiente del voltaje. El maltrato infantil contribuyó al estado de metilación de CpG2 III de la 5-hidroxitriptamina 3A, lo que altera la forma en que el maltrato afecta al trastorno bipolar.
Además, las intervenciones terapéuticas, como los factores de transcripción modificados , podrían modificar la estructura de la cromatina para abordar los cambios epigenéticos que se encuentran en las personas con trastorno bipolar. Los inhibidores de la ADN metiltransferasa (DNMT) y los inhibidores de la histona desacetilasa (HDAC) podrían revertir las modificaciones epigenéticas para abordar terapéuticamente el trastorno bipolar. Los inhibidores de la DNMT y la HDAC a menudo producen efectos similares a los antidepresivos.