Boron, Walter F.; Boulpaep, Emile L., eds. (2017). Fisiología médica (3.ª ed.). Filadelfia, PA: Elsevier. ISBN 978-1-4557-4377-3.

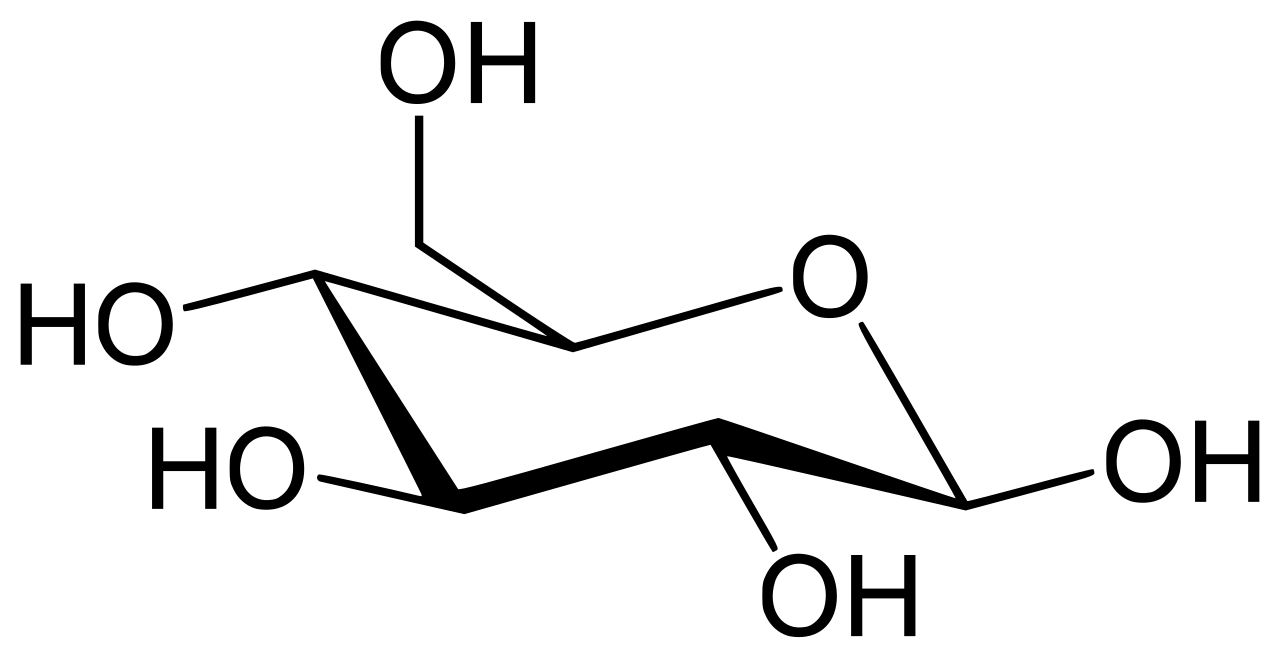
La enzima glucosa 6-fosfatasa (EC 3.1.3.9, G6Pasa ; nombre sistemático D -glucosa-6-fosfato fosfohidrolasa ) cataliza la hidrólisis de la glucosa 6-fosfato , lo que da como resultado la creación de un grupo fosfato y glucosa libre:
Durante el ayuno, los niveles adecuados de glucosa en sangre están asegurados por la glucosa liberada de los depósitos de glucógeno hepáticos mediante glucogenólisis , así como por la glucosa generada por gluconeogénesis en el hígado y, en menor medida, en los riñones. La G6P es el producto de ambas vías [1] y debe convertirse en glucosa antes de que pueda ser exportada desde la célula a la sangre por los transportadores de glucosa unidos a la membrana . [2] Por lo tanto, la G6Pasa se expresa principalmente en el hígado y el riñón [1] ; aunque el músculo esquelético contiene colectivamente la reserva de glucógeno más sustancial del cuerpo, la glucosa no puede movilizarse desde él ya que el músculo carece de G6Pasa. [3] : 1171
La insulina inhibe la actividad hepática de la G6Pasa, [3] : 1046 mientras que el glucagón la promueve. [3] : 1052 La expresión de la G6Pasa aumenta durante la inanición, en la diabetes y por los glucocorticosteroides . [1]
La glucosa 6-fosfatasa es un complejo de múltiples proteínas componentes, que incluyen transportadores de G6P, glucosa y fosfato. La función principal de la fosfatasa la realiza la subunidad catalítica de la glucosa 6-fosfatasa. En los seres humanos, existen tres isoenzimas de la subunidad catalítica: glucosa 6-fosfatasa-α, codificada por G6PC ; IGRP, codificada por G6PC2 ; y glucosa 6-fosfatasa-β, codificada por G6PC3 . [4]
La glucosa 6-fosfatasa-α y la glucosa 6-fosfatasa-β son fosfohidrolasas funcionales y tienen una estructura de sitio activo, topología, mecanismo de acción y propiedades cinéticas similares con respecto a la hidrólisis de G6P. [5] Por el contrario, la IGRP casi no tiene actividad hidrolasa y puede desempeñar un papel diferente en la estimulación de la secreción de insulina pancreática. [6]
.png/1280px-Wiki_image_(2).png)
.png/1280px-Final_for_wiki_(1).png)

Aunque no se ha llegado a un consenso claro, un gran número de científicos se adhieren a un modelo de transporte de sustrato para explicar las propiedades catalíticas de la glucosa 6-fosfatasa. En este modelo, la glucosa 6-fosfatasa tiene un bajo grado de selectividad. La transferencia de la glucosa 6-fosfato se lleva a cabo por una proteína transportadora (T1) y el retículo endoplasmático (RE) contiene estructuras que permiten la salida del grupo fosfato (T2) y la glucosa (T3). [7]
La glucosa 6-fosfatasa consta de 357 aminoácidos y está anclada al retículo endoplasmático (RE) mediante nueve hélices transmembrana. Su extremo N -terminal y su sitio activo se encuentran en el lado del lumen del RE y su extremo C -terminal se proyecta hacia el citoplasma. Debido a su estrecha asociación con el RE, la estructura exacta de la glucosa 6-fosfatasa sigue siendo desconocida. Sin embargo, la alineación de secuencias ha demostrado que la glucosa 6-fosfatasa es estructuralmente similar al sitio activo de la cloroperoxidasa que contiene vanadio que se encuentra en Curvularia inaequalis. [8]
Basándose en estudios cinéticos de pH de la catálisis de la glucosa 6-fosfatasa-α, se propuso que la hidrólisis de la glucosa 6-fosfato se completaba a través de un intermediario covalente de glucosa 6-fosfato de fosfohistidina. El sitio activo de la glucosa 6-fosfatasa-α se identificó inicialmente por la presencia de un motivo característico de fosfato conservado que se encuentra habitualmente en las fosfatasas lipídicas, las fosfatasas ácidas y las haloperoxidasas de vanadio. [5]
Los residuos esenciales en el sitio activo de las haloperoxidasas de vanadio incluyen: Lys353, Arg360, Arg490, His404 e His496. Los residuos correspondientes en el sitio activo de la glucosa 6-fosfatasa-α incluyen Arg170 y Arg83, que donan iones de hidrógeno al fosfato, estabilizando el estado de transición, His119, que proporciona un protón al oxígeno desfosforilado unido a la glucosa, e His176, que completa un ataque nucleofílico sobre el fosfato para formar un intermedio enzimático de fosforilo unido covalentemente. [9] Dentro de la cloroperoxidasa que contiene vanadio, se encontró que Lys353 estabilizaba el fosfato en el estado de transición. Sin embargo, el residuo correspondiente en la glucosa 6-fosfatasa-α (Lys76) reside dentro de la membrana del RE y su función, si la hay, actualmente no se ha determinado. Con excepción de Lys76, todos estos residuos se encuentran en el lado luminal de la membrana del RE. [5]
La glucosa 6-fosfatasa-β es una proteína de membrana de 346 aminoácidos que se expresa de forma ubicua y que comparte un 36 % de identidad de secuencia con la glucosa 6-fosfatasa-α. Dentro de la enzima glucosa 6-fosfatasa-β, las alineaciones de secuencia predicen que su sitio activo contiene His167, His114 y Arg79. De manera similar al sitio activo de la glucosa 6-fosfatasa-α, His167 es el residuo que proporciona el ataque nucleofílico, y His114 y Arg79 son los donantes de hidrógeno. La glucosa 6-fosfatasa-β también se localiza en la membrana del RE, aunque se desconoce su orientación. [5]
La hidrólisis de la glucosa 6-fosfato comienza con un ataque nucleofílico sobre el fosfato unido al azúcar por parte de His176, lo que da como resultado la formación de un enlace de fosfohistidina y la degradación de un carbonilo. A continuación, un oxígeno cargado negativamente transfiere sus electrones, reformando un carbonilo y rompiendo su enlace con la glucosa. El oxígeno cargado negativamente unido a la glucosa es protonado por His119, formando una glucosa libre. El intermediario fosfo producido por la reacción entre His176 y el grupo fosfato se rompe luego por un ataque hidrófilo; después de la adición de otro hidróxido y la descomposición de un carbonilo, el carbonilo se reforma, lo que libera los electrones donados originalmente por el residuo de His176, creando así un grupo fosfato libre y completando la hidrólisis. [9]
Los genes que codifican la enzima se expresan principalmente en el hígado, en la corteza renal y (en menor medida) en las células β de los islotes pancreáticos y la mucosa intestinal (especialmente durante períodos de inanición). [7] La glucosa 6-fosfatasa está presente en una amplia variedad de músculos en todo el reino animal, aunque en concentraciones muy bajas. [10] Por lo tanto, el glucógeno que almacenan los músculos no suele estar disponible para el resto de las células del cuerpo porque la glucosa 6-fosfato no puede atravesar el sarcolema a menos que se desfosforile. La enzima desempeña un papel importante durante los períodos de ayuno y cuando los niveles de glucosa son bajos. Se ha demostrado que la inanición y la diabetes inducen un aumento de dos a tres veces en la actividad de la glucosa 6-fosfatasa en el hígado. [7] La actividad de la Glc 6-Pasa también aumenta drásticamente al nacer cuando un organismo se independiza de la fuente de glucosa de la madre. El gen humano Glc 6-Pase contiene cinco exones que abarcan aproximadamente 125,5 kb de ADN ubicado en el cromosoma 17q21. [11]
Las mutaciones del sistema de la glucosa 6-fosfatasa, específicamente las subunidades de la glucosa 6-fosfatasa-α (glucosa 6-fosfatasa-α), del transportador de glucosa 6 (G6PT) y de la glucosa 6-fosfatasa-β (glucosa 6-fosfatasa-β o G6PC3), conducen a deficiencias en el mantenimiento de la homeostasis de la glucosa interprandial y de la función y homeostasis de los neutrófilos . [12] [13] Las mutaciones tanto en la glucosa 6-fosfatasa-α como en la G6PT conducen a la enfermedad de almacenamiento de glucógeno tipo I (GSD 1, enfermedad de von Gierke). [14] Para ser más específicos, las mutaciones en la glucosa-6-fosfatasa-α conducen a la enfermedad de almacenamiento de glucógeno tipo 1a, que se caracteriza por la acumulación de glucógeno y grasa en el hígado y los riñones, lo que resulta en hepatomegalia y renomegalia. [15] La GSD-1a constituye aproximadamente el 80% de los casos de GSD-1 que se presentan clínicamente. [16] La ausencia de G6PT conduce a la GSD-1b (GSD-1b), que se caracteriza por la falta de un G6PT y representa el 20% de los casos que se presentan clínicamente. [16] [17]

La causa específica de la GSD-1a se origina de mutaciones sin sentido, inserciones/deleciones con o sin un cambio en el marco de lectura, o mutaciones del sitio de empalme que ocurren a nivel genético. [7] Las mutaciones sin sentido afectan los dos grandes bucles luminales y hélices transmembrana de la glucosa 6-fosfatasa-α, eliminando o reduciendo en gran medida la actividad de la enzima. [7] La causa específica de la GSD-1b se origina de mutaciones "graves" como mutaciones del sitio de empalme, mutaciones de cambio de marco y sustituciones de un residuo altamente conservado que destruyó completamente la actividad de G6PT. [7] Estas mutaciones conducen a la prevalencia de la GSD-1 al prevenir el transporte de glucosa-6-fosfato (G6P) a la porción luminal del RE y también inhibir la conversión de G6P en glucosa para ser utilizada por la célula.
El tercer tipo de deficiencia de glucosa 6-fosfatasa, la deficiencia de glucosa 6-fosfatasa-β, se caracteriza por un síndrome de neutropenia congénita en el que los neutrófilos muestran un estrés aumentado del retículo endoplasmático (RE), aumento de la apoptosis, deterioro de la homeostasis energética y deterioro de la funcionalidad. [18] También puede conducir a malformaciones cardíacas y urogenitales. [19] Esta tercera clase de deficiencia también se ve afectada por una deficiencia de G6PT, ya que la glucosa-6-fosfatasa-β también se encuentra dentro del lumen del RE y, por lo tanto, puede conducir a síntomas similares de deficiencia de glucosa-6-fosfatasa-β asociados con GSD-1b. [17] Además, estudios recientes han dilucidado esta área de similitud entre ambas deficiencias y han demostrado que la glicosilación aberrante ocurre en ambas deficiencias. [20] La glicosilación de los neutrófilos tiene un efecto profundo en la actividad de los neutrófilos y, por lo tanto, también puede clasificarse como un trastorno de glicosilación congénita. [20]
Se ha determinado que la función principal de la glucosa 6-fosfatasa-β es proporcionar glucosa reciclada al citoplasma de los neutrófilos para mantener su función normal. La alteración de la relación glucosa/G6P debido a una disminución significativa de los niveles de glucosa intracelular provoca una alteración significativa de la glucólisis y el HMS . [13] A menos que se contrarreste mediante la captación de glucosa extracelular, esta deficiencia conduce a una disfunción de los neutrófilos. [13]
Se ha demostrado que los compuestos de vanadio, como el sulfato de vanadilo , inhiben la enzima y, por lo tanto, aumentan la sensibilidad a la insulina in vivo en diabéticos, según se evaluó mediante la técnica de pinza hiperinsulinémica , lo que puede tener posibles implicaciones terapéuticas. [21] [22]
Las imágenes de gráficos moleculares se produjeron utilizando UCSF Chimera. [23]
{{cite journal}}: Requiere citar revista |journal=( ayuda ){kind=link}