Tumor de Wilms
[2] Toma el nombre de Max Wilms (1867-1918), cirujano alemán que lo identificó por primera vez.
Se ubican con más frecuencia en el polo superior del riñón; tienden a ser tumores encapsulados y vascularizados que no rebasan la línea media hacia el lado opuesto del abdomen.
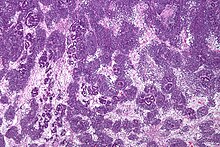
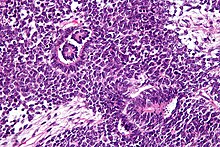
Es característica su presencia en los glomérulos y túbulos renales rodeado por un estroma celular.
[4] Los cambios en este cromosoma predisponen al portador a un alto riesgo de trastornos congénitos, incluyendo el nefroblastoma.
Si el ultrasonido no esclarece el origen de la lesión, se sugiere correlacionar con una tomografía.
Primero es obligatorio conocer la extensión de la enfermedad para optimizar el plan terapéutico.
La extirpación quirúrgica del tumor es necesaria, así como otros tejidos adyacentes que puedan estar afectados.
La sobrevivencia al cabo de cuatro años es de 94 % para aquellos pacientes cuya lesión no es más avanzada que una fase I o fase II, 76 % para aquellos cuya lesión más avanzada era la etapa III.