En química , una molécula hipervalente (el fenómeno a veces se conoce coloquialmente como octeto expandido ) es una molécula que contiene uno o más elementos del grupo principal que aparentemente tienen más de ocho electrones en sus capas de valencia . El pentacloruro de fósforo ( PCl 5 ), el hexafluoruro de azufre ( SF 6 ), el trifluoruro de cloro ( ClF 3 ), el clorito ( ClO−2) en ácido cloroso y el triyoduro ( I−3) Los iones son ejemplos de moléculas hipervalentes.
Las moléculas hipervalentes fueron definidas formalmente por primera vez por Jeremy I. Musher en 1969 como moléculas que tienen átomos centrales del grupo 15-18 en cualquier valencia distinta a la más baja (es decir, 3, 2, 1, 0 para los grupos 15, 16, 17, 18 respectivamente, según la regla del octeto ). [1]
Existen varias clases específicas de moléculas hipervalentes:
La nomenclatura NXL, introducida en colaboración por los grupos de investigación de Martin , Arduengo y Kochi en 1980, [2] se utiliza a menudo para clasificar los compuestos hipervalentes de los elementos del grupo principal, donde:
Algunos ejemplos de nomenclatura NXL incluyen:
El debate sobre la naturaleza y clasificación de las moléculas hipervalentes se remonta a Gilbert N. Lewis e Irving Langmuir y al debate sobre la naturaleza del enlace químico en la década de 1920. [3] Lewis mantuvo la importancia del enlace de dos centros y dos electrones (2c-2e) para describir la hipervalencia, por lo que utilizó octetos expandidos para explicar dichas moléculas. Utilizando el lenguaje de la hibridación orbital, se decía que los enlaces de moléculas como PF 5 y SF 6 se construían a partir de orbitales sp 3 d n en el átomo central. Langmuir, por otro lado, defendió el predominio de la regla del octeto y prefirió el uso de enlaces iónicos para explicar la hipervalencia sin violar la regla (por ejemplo, " SF2+
42F − " para SF 6 ).
A finales de los años 1920 y 1930, Sugden defendió la existencia de un enlace de un electrón y dos centros (2c-1e) y, por tanto, racionalizó la unión en moléculas hipervalentes sin necesidad de octetos expandidos o carácter de enlace iónico; esto fue poco aceptado en ese momento. [3] En los años 1940 y 1950, Rundle y Pimentel popularizaron la idea del enlace de cuatro electrones y tres centros , que es esencialmente el mismo concepto que Sugden intentó proponer décadas antes; el enlace de cuatro electrones y tres centros puede verse alternativamente como si constara de dos enlaces de un electrón y dos centros colineales, con los dos electrones no enlazantes restantes localizados en los ligandos. [3]
El intento de preparar moléculas orgánicas hipervalentes comenzó con Hermann Staudinger y Georg Wittig en la primera mitad del siglo XX, quienes buscaron desafiar la teoría de valencia existente y preparar con éxito moléculas hipervalentes centradas en nitrógeno y fósforo. [4] La base teórica para la hipervalencia no fue delineada hasta el trabajo de JI Musher en 1969. [1]
En 1990, Magnusson publicó un trabajo seminal que excluía definitivamente la importancia de la hibridación del orbital d en el enlace de compuestos hipervalentes de elementos de segunda fila. Esto había sido durante mucho tiempo un punto de controversia y confusión al describir estas moléculas utilizando la teoría de orbitales moleculares . Parte de la confusión aquí se origina del hecho de que uno debe incluir funciones d en los conjuntos de base utilizados para describir estos compuestos (o de lo contrario resultan energías irrazonablemente altas y geometrías distorsionadas), y la contribución de la función d a la función de onda molecular es grande. Estos hechos se interpretaron históricamente como que significaban que los orbitales d deben estar involucrados en el enlace. Sin embargo, Magnusson concluye en su trabajo que la participación del orbital d no está implicada en la hipervalencia. [5]
Sin embargo, un estudio de 2013 mostró que, aunque el modelo iónico de Pimentel explica mejor el enlace de especies hipervalentes, la contribución energética de una estructura de octeto expandida tampoco es nula. En este estudio de la teoría del enlace de valencia moderna del enlace del difluoruro de xenón , se encontró que las estructuras iónicas representan alrededor del 81% de la función de onda general, de la cual el 70% surge de estructuras iónicas que emplean solo el orbital p en xenón, mientras que el 11% surge de estructuras iónicas que emplean un híbrido en xenón. La contribución de una estructura formalmente hipervalente que emplea un orbital de hibridación sp 3 d en xenón representa el 11% de la función de onda, y una contribución dirradical constituye el 8% restante. La contribución del 11% sp 3 d da como resultado una estabilización neta de la molécula de 7,2 kcal (30 kJ) mol −1 , [6] una fracción menor pero significativa de la energía total de la energía de enlace total (64 kcal (270 kJ) mol −1 ). [7] Otros estudios han encontrado de manera similar contribuciones energéticas menores pero no despreciables de las estructuras de octetos expandidos en SF 6 (17%) y XeF 6 (14%). [8]
A pesar de la falta de realismo químico, la IUPAC recomienda el dibujo de estructuras de octetos expandidos para grupos funcionales como sulfonas y fosforanos , con el fin de evitar el dibujo de una gran cantidad de cargas formales o enlaces simples parciales. [9]
Un tipo especial de moléculas hipervalentes son los hidruros hipervalentes. La mayoría de las moléculas hipervalentes conocidas contienen sustituyentes más electronegativos que sus átomos centrales. [10] [11] Los hidruros hipervalentes son de especial interés porque el hidrógeno suele ser menos electronegativo que el átomo central. Se han realizado varios estudios computacionales sobre hidruros de calcógeno [11] [12] [13] [14] [15] [16] e hidruros de pnicógeno . [17] [18] [19] [20] [21] Recientemente, un nuevo estudio computacional ha demostrado que la mayoría de los hidruros de halógeno hipervalentes XH n pueden existir. Se sugiere que IH 3 e IH 5 son lo suficientemente estables como para ser observables o, posiblemente, incluso aislables. [22]
Tanto el término como el concepto de hipervalencia siguen siendo objeto de críticas. En 1984, en respuesta a esta controversia general, Paul von Ragué Schleyer propuso sustituir el término «hipervalencia» por el de hipercoordinación , ya que este término no implica ningún modo de enlace químico y, por tanto, la cuestión podía evitarse por completo. [3]
El concepto en sí ha sido criticado por Ronald Gillespie quien, basándose en un análisis de las funciones de localización de electrones, escribió en 2002 que "como no hay una diferencia fundamental entre los enlaces en moléculas hipervalentes y no hipervalentes (octeto de Lewis), no hay razón para seguir usando el término hipervalente". [23]
En el caso de moléculas hipercoordinadas con ligandos electronegativos como el PF 5 , se ha demostrado que los ligandos pueden extraer suficiente densidad electrónica del átomo central para que su contenido neto sea nuevamente de 8 electrones o menos. En consonancia con esta visión alternativa está el hallazgo de que las moléculas hipercoordinadas basadas en ligandos de flúor, por ejemplo el PF 5 , no tienen homólogos hidruros , por ejemplo el fosforano (PH 5 ), que es desconocido.
El modelo iónico se sostiene bien en los cálculos termoquímicos y predice una formación exotérmica favorable de PF .+
4F−
a partir de trifluoruro de fósforo PF 3 y flúor F 2 mientras que una reacción similar forma PH+
4yo−
no es favorable. [24]
Durrant ha propuesto una definición alternativa de hipervalencia, basada en el análisis de mapas de carga atómica obtenidos a partir de átomos en la teoría de moléculas. [25] Este enfoque define un parámetro llamado equivalente electrónico de valencia, γ, como “el recuento formal de electrones compartidos en un átomo dado, obtenido por cualquier combinación de formas de resonancia iónica y covalente válidas que reproduce la distribución de carga observada”. Para cualquier átomo X en particular, si el valor de γ(X) es mayor que 8, ese átomo es hipervalente. Usando esta definición alternativa, muchas especies como PCl 5 , SO2−
4, y XeF 4 , que son hipervalentes según la definición de Musher, se reclasifican como hipercoordinados pero no hipervalentes, debido a un fuerte enlace iónico que aleja a los electrones del átomo central. Por otro lado, algunos compuestos que normalmente se escriben con enlaces iónicos para cumplir con la regla del octeto, como el ozono O 3 , el óxido nitroso NNO y el N-óxido de trimetilamina (CH
3)
3NO , se ha demostrado que son realmente hipervalentes. Ejemplos de cálculos de γ para fosfato PO3−
4(γ(P) = 2,6, no hipervalente) y ortonitrato NO3−
4(γ(N) = 8,5, hipervalente) se muestran a continuación.

Las primeras consideraciones sobre la geometría de las moléculas hipervalentes devolvieron disposiciones familiares que se explicaban bien mediante el modelo VSEPR para el enlace atómico. En consecuencia, las moléculas de tipo AB 5 y AB 6 poseerían una geometría trigonal bipiramidal y octaédrica, respectivamente. Sin embargo, para tener en cuenta los ángulos de enlace observados, las longitudes de enlace y la aparente violación de la regla del octeto de Lewis , se han propuesto varios modelos alternativos.
En la década de 1950 se propuso un tratamiento de capa de valencia expandida del enlace hipervalente para explicar la arquitectura molecular, donde el átomo central de las moléculas penta- y hexacoordinadas utilizaría AO d además de AO s y p. Sin embargo, los avances en el estudio de los cálculos ab initio han revelado que la contribución de los orbitales d al enlace hipervalente es demasiado pequeña para describir las propiedades de enlace, y esta descripción ahora se considera mucho menos importante. [5] Se demostró que en el caso del SF 6 hexacoordinado , los orbitales d no están involucrados en la formación del enlace SF, pero la transferencia de carga entre los átomos de azufre y flúor y las estructuras de resonancia apropiadas pudieron explicar la hipervalencia (ver a continuación).
Se han intentado modificaciones adicionales a la regla del octeto para involucrar características iónicas en el enlace hipervalente. Como una de estas modificaciones, en 1951, se propuso el concepto de enlace de 3 centros y 4 electrones (3c-4e) , que describía el enlace hipervalente con un orbital molecular cualitativo . El enlace 3c-4e se describe como tres orbitales moleculares dados por la combinación de un orbital atómico p en el átomo central y un orbital atómico de cada uno de los dos ligandos en lados opuestos del átomo central. Solo uno de los dos pares de electrones ocupa un orbital molecular que implica un enlace al átomo central, siendo el segundo par no enlazante y ocupando un orbital molecular compuesto solo por orbitales atómicos de los dos ligandos. Este modelo en el que se conserva la regla del octeto también fue defendido por Musher. [3]

Una descripción completa de las moléculas hipervalentes surge de la consideración de la teoría de orbitales moleculares a través de métodos mecánicos cuánticos. Un LCAO en, por ejemplo, hexafluoruro de azufre, tomando un conjunto base de un orbital 3s de azufre, los tres orbitales 3p de azufre y seis combinaciones lineales adaptadas a la simetría de geometría octaédrica (SALC) de orbitales de flúor, se obtiene un total de diez orbitales moleculares (cuatro OM de enlace completamente ocupados de la energía más baja, dos OM no enlazantes de energía intermedia completamente ocupados y cuatro OM antienlazantes vacantes con la energía más alta) que brindan espacio para los 12 electrones de valencia. Esta es una configuración estable solo para moléculas S X 6 que contienen átomos de ligando electronegativos como el flúor, lo que explica por qué SH 6 no es una molécula estable. En el modelo de enlace, los dos OM no enlazantes (1e g ) están localizados por igual en los seis átomos de flúor.
Para compuestos hipervalentes en los que los ligandos son más electronegativos que el átomo central hipervalente, se pueden dibujar estructuras de resonancia con no más de cuatro enlaces de pares de electrones covalentes y completarse con enlaces iónicos para obedecer la regla del octeto. Por ejemplo, en el pentafluoruro de fósforo (PF 5 ), se pueden generar 5 estructuras de resonancia cada una con cuatro enlaces covalentes y un enlace iónico con mayor peso en las estructuras colocando el carácter iónico en los enlaces axiales, satisfaciendo así la regla del octeto y explicando tanto la geometría molecular bipiramidal trigonal observada como el hecho de que la longitud del enlace axial (158 pm) es mayor que la ecuatorial (154 pm). [26]
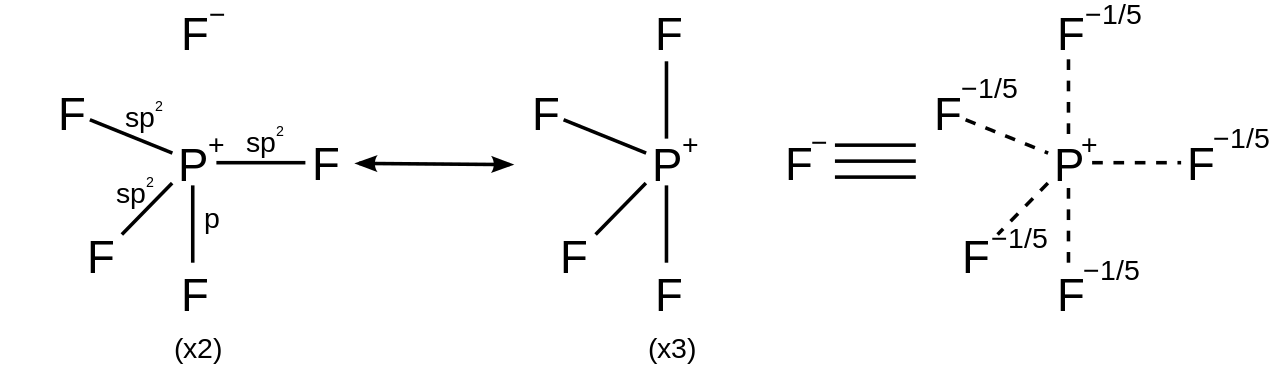
En el caso de una molécula hexacoordinada, como el hexafluoruro de azufre , cada uno de los seis enlaces tiene la misma longitud. La racionalización descrita anteriormente se puede aplicar para generar 15 estructuras de resonancia, cada una con cuatro enlaces covalentes y dos enlaces iónicos, de modo que el carácter iónico se distribuya de manera uniforme en cada uno de los enlaces azufre-flúor.

La teoría del enlace de valencia acoplado a espín se ha aplicado al diazometano y el análisis orbital resultante se interpretó en términos de una estructura química en la que el nitrógeno central tiene cinco enlaces covalentes;

Esto llevó a los autores a la interesante conclusión de que "contrariamente a lo que nos enseñaron a todos cuando éramos estudiantes, el átomo de nitrógeno de hecho forma cinco enlaces covalentes y la disponibilidad o no de orbitales d no tiene nada que ver con este estado de cosas". [27]
Las moléculas de fósforo hexacoordinadas que involucran ligandos de nitrógeno, oxígeno o azufre proporcionan ejemplos de hexacoordinación de ácido de Lewis-base de Lewis. [28] Para los dos complejos similares que se muestran a continuación, la longitud del enlace C–P aumenta con la disminución de la longitud del enlace N–P; la fuerza del enlace C–P disminuye con el aumento de la fuerza de la interacción ácido de Lewis–base de Lewis N–P.

Esta tendencia también es generalmente cierta para los elementos del grupo principal pentacoordinados con uno o más ligandos que contienen pares solitarios, incluidos los ejemplos de silicio pentacoordinados con oxígeno que se muestran a continuación.

Los enlaces Si-halógeno varían desde cerca del valor de van der Waals esperado en A (un enlace débil) hasta casi el valor de enlace simple covalente esperado en C (un enlace fuerte). [28]
Corriu y colaboradores realizaron un trabajo inicial que caracterizaba las reacciones que se pensaba que se desarrollaban a través de un estado de transición hipervalente. [29] Las mediciones de las velocidades de reacción de hidrólisis de clorosilanos tetravalentes incubados con cantidades catalíticas de agua arrojaron una velocidad que es de primer orden en clorosilano y de segundo orden en agua. Esto indicó que dos moléculas de agua interactuaron con el silano durante la hidrólisis y a partir de esto se propuso un mecanismo de reacción binucleofílica. Corriu y colaboradores luego midieron las velocidades de hidrólisis en presencia del catalizador nucleofílico HMPT, DMSO o DMF. Se demostró que la velocidad de hidrólisis era nuevamente de primer orden en clorosilano, de primer orden en catalizador y ahora de primer orden en agua. Apropiadamente, las velocidades de hidrólisis también exhibieron una dependencia de la magnitud de la carga en el oxígeno del nucleófilo.
En conjunto, todo esto llevó al grupo a proponer un mecanismo de reacción en el que se produce un ataque nucleofílico determinante de la velocidad del silano tetracoordinado por el nucleófilo (o agua) en el que se forma un silano pentacoordinado hipervalente. A esto le sigue un ataque nucleofílico del intermediario por el agua en un paso determinante de la velocidad que conduce a especies hexacoordinadas que se descomponen rápidamente dando lugar al hidroxisilano.
Holmes y sus colaboradores [30] investigaron más a fondo la hidrólisis del silano, en la que se encontró que el Mes tetracoordinado
2SiF
2(Mes = mesitilo ) y Mes pentacoordinado
2SiF−
3se hicieron reaccionar con dos equivalentes de agua. Después de veinticuatro horas, casi no se observó hidrólisis del silano tetracoordinado, mientras que el silano pentacoordinado se hidrolizó por completo después de quince minutos. Además, los datos de difracción de rayos X recopilados para las sales de tetraetilamonio de los fluorosilanos mostraron la formación de una red de bisilonato de hidrógeno que soporta un intermedio hexacoordinado del que se obtiene HF−
2se desplaza rápidamente dando lugar al producto hidroxilado. Esta reacción y los datos cristalográficos respaldan el mecanismo propuesto por Corriu et al .

También se ha observado una reactividad aparentemente mayor de las moléculas hipervalentes, en contraste con los análogos tetravalentes, en las reacciones de Grignard. El grupo Corriu midió [31] los tiempos medios de la reacción de Grignard mediante RMN para sales de potasio 18-corona-6 relacionadas de una variedad de fluorosilanos tetra- y pentacoordinados en presencia de cantidades catalíticas de nucleófilo.
Aunque el método de semirreacción es impreciso, las diferencias de magnitud en las velocidades de reacción permitieron un esquema de reacción propuesto en el que un ataque previo a la determinación de la velocidad del silano tetravalente por parte del nucleófilo da como resultado un equilibrio entre las especies tetracoordinadas neutras y el compuesto pentavalente aniónico. A esto le sigue la coordinación nucleofílica por parte de dos reactivos de Grignard, como se observa normalmente, formando un estado de transición hexacoordinado y obteniendo el producto esperado.

Las implicaciones mecanicistas de esto se extienden a una especie de silicio hexacoordinado que se cree que es activa como estado de transición en algunas reacciones. La reacción de los trifluorosilanos alílicos o crotílicos con aldehídos y cetonas solo precede con la activación del fluoruro para dar un silicio pentacoordinado. Este intermediario actúa entonces como un ácido de Lewis para coordinarse con el átomo de oxígeno del carbonilo. El debilitamiento adicional del enlace silicio-carbono a medida que el silicio se vuelve hexacoordinado ayuda a impulsar esta reacción. [32]

También se ha observado una reactividad similar para otras estructuras hipervalentes, como la mezcla de compuestos de fósforo, para los que se han propuesto estados de transición hexacoordinados. Se ha estudiado la hidrólisis de fosforanos y oxifosforanos [33] y se ha demostrado que es de segundo orden en agua. Bel'skii et al . han propuesto un ataque nucleofílico determinante de la velocidad por agua que da como resultado un equilibrio entre las especies de fósforo penta- y hexacoordinadas, que es seguido por una transferencia de protones que involucra a la segunda molécula de agua en un paso de apertura del anillo que determina la velocidad, lo que conduce al producto hidroxilado.

También se ha postulado que la alcoholisis de compuestos de fósforo pentacoordinados, como el trimetoxifosfoleno con alcohol bencílico, ocurre a través de un estado de transición octaédrico similar, como en la hidrólisis, pero sin apertura del anillo. [34]

A partir de estos experimentos se puede entender que la mayor reactividad observada para las moléculas hipervalentes, en contraste con compuestos análogos no hipervalentes, se puede atribuir a la congruencia de estas especies con los estados activados hipercoordinados que normalmente se forman durante el curso de la reacción.
La reactividad mejorada en el silicio pentacoordinado no se entiende completamente. Corriu y colaboradores sugirieron que un mayor carácter electropositivo en el átomo de silicio pentavalente puede ser responsable de su mayor reactividad. [35] Los cálculos preliminares ab initio respaldaron esta hipótesis hasta cierto punto, pero utilizaron un conjunto de base pequeño. [36]
Dieters y colaboradores utilizaron un programa informático para realizar cálculos ab initio, Gaussian 86 , para comparar el silicio y el fósforo tetracoordinados con sus análogos pentacoordinados. Este enfoque ab initio se utiliza como complemento para determinar por qué la reactividad mejora en las reacciones nucleofílicas con compuestos pentacoordinados. Para el silicio, se utilizó el conjunto de bases 6-31+G* debido a su carácter aniónico pentacoordinado y, para el fósforo, se utilizó el conjunto de bases 6-31G* . [36]
Los compuestos pentacoordinados deberían ser teóricamente menos electrofílicos que los análogos tetracoordinados debido al impedimento estérico y a la mayor densidad electrónica de los ligandos, pero experimentalmente muestran una mayor reactividad con los nucleófilos que sus análogos tetracoordinados. Se realizaron cálculos ab initio avanzados en series de especies tetracoordinadas y pentacoordinadas para comprender mejor este fenómeno de reactividad. Cada serie varió según el grado de fluoración. Las longitudes de enlace y las densidades de carga se muestran como funciones de cuántos ligandos hidruro hay en los átomos centrales. Por cada nuevo hidruro, hay un fluoruro menos. [36]
Para las longitudes de enlace de silicio y fósforo, las densidades de carga y la superposición de enlaces de Mulliken, se calcularon poblaciones para especies tetra y pentacoordinadas mediante este enfoque ab initio. [36] La adición de un ion fluoruro al silicio tetracoordinado muestra un aumento promedio general de 0,1 carga electrónica, que se considera insignificante. En general, las longitudes de enlace en especies pentacoordinadas bipiramidales trigonales son más largas que las de los análogos tetracoordinados. Los enlaces Si-F y Si-H aumentan en longitud tras la pentacoordinación y se observan efectos relacionados en las especies de fósforo, pero en menor grado. La razón de la mayor magnitud en el cambio de longitud de enlace para las especies de silicio sobre las especies de fósforo es el aumento de la carga nuclear efectiva en el fósforo. Por lo tanto, se concluye que el silicio está unido de forma más débil a sus ligandos.
Además, Dieters y colaboradores [36] muestran una correlación inversa entre la longitud de enlace y la superposición de enlaces para todas las series. Se concluye que las especies pentacoordinadas son más reactivas debido a sus enlaces más laxos como estructuras trigono-bipiramidales.
Al calcular las energías para la adición y eliminación de un ion fluoruro en varias especies de silicio y fósforo, se encontraron varias tendencias. En particular, las especies tetracoordinadas tienen requerimientos de energía mucho mayores para la eliminación de ligando que las especies pentacoordinadas. Además, las especies de silicio tienen requerimientos de energía menores para la eliminación de ligando que las especies de fósforo, lo que es un indicio de enlaces más débiles en el silicio.
{{cite book}}: CS1 maint: varios nombres: lista de autores ( enlace ){{cite journal}}: Falta o está vacío |title=( ayuda )