En cuanto al mecanismo, hay tres genes, GP1BA , GP1BB y GP9 que están involucrados (debido a mutaciones). [3] Estas mutaciones no permiten que el complejo GPIb-IX-V se una al factor von Willebrand, que a su vez es lo que ayudaría a que las plaquetas se adhieran a un sitio de lesión, lo que eventualmente ayuda a detener el sangrado. [2]
Diagnóstico
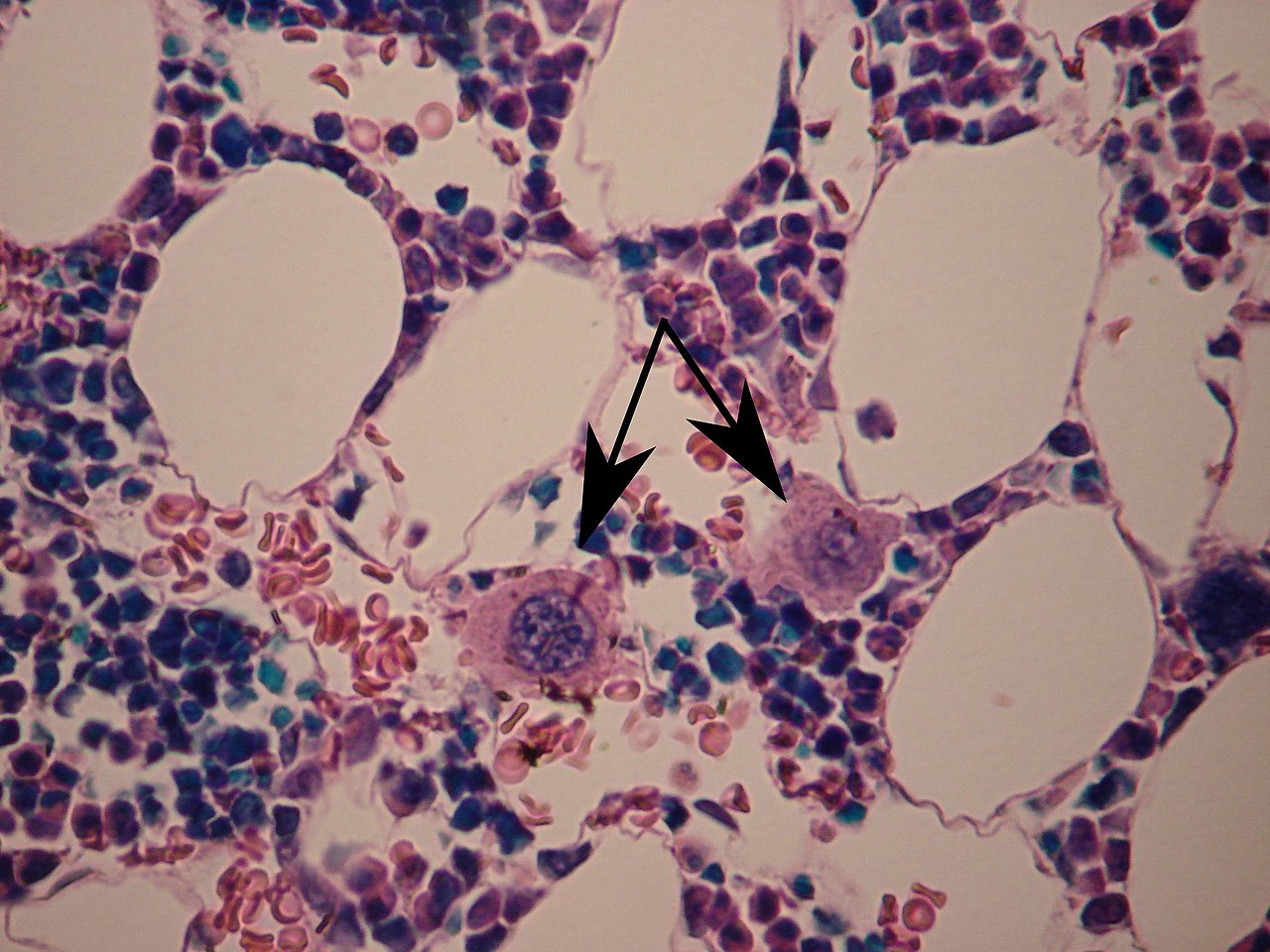
Megacariocitos (flechas)
En términos de diagnóstico, el síndrome de Bernard-Soulier se caracteriza por un tiempo de sangrado prolongado, trombocitopenia , aumento de megacariocitos y plaquetas agrandadas. El síndrome de Bernard-Soulier se asocia con defectos cuantitativos o cualitativos del complejo de glucoproteína plaquetaria GPIb/V/IX. El grado de trombocitopenia puede estimarse incorrectamente, debido a la posibilidad de que cuando el recuento de plaquetas se realiza con contadores automáticos, las plaquetas gigantes puedan alcanzar el tamaño de los glóbulos rojos. Las plaquetas grandes y el bajo recuento de plaquetas en el SBS aparentemente se deben a la ausencia de GPIbα y del sitio de unión de la filamina A que une el complejo GPIb-IX-V al esqueleto de la membrana plaquetaria. [5] [8]
Diagnóstico diferencial
El diagnóstico diferencial del síndrome de Bernard-Soulier incluye tanto la trombastenia de Glanzmann como la enfermedad de von Willebrand pediátrica. [5] Las plaquetas del síndrome de Bernard-Soulier no se agregan a la ristocetina y este defecto no se corrige con la adición de plasma normal, lo que lo distingue de la enfermedad de von Willebrand. [4] A continuación se muestra una tabla que compara su resultado con otros trastornos de agregación plaquetaria:
Tratamiento
Ácido tranexámico
Los episodios de sangrado pueden controlarse mediante transfusión de plaquetas. La mayoría de los heterocigotos, con pocas excepciones, no presentan diátesis hemorrágica . El síndrome de Behrens-Schneider se presenta como un trastorno hemorrágico debido a la incapacidad de las plaquetas de unirse y agregarse en los sitios de lesión endotelial vascular. [4] En caso de sangrado de las mucosas, se puede administrar ácido tranexámico . [5]
La frecuencia del síndrome de Bernard-Soulier es de aproximadamente 1 en 1.000.000 de personas. [11] El síndrome, identificado en el año 1948, lleva el nombre del Dr. Jean Bernard y el Dr. Jean Pierre Soulier . [12]
^ ab Lanza F (2006). "Síndrome de Bernard-Soulier (distrofia trombocítica hemorrágica)". Orphanet J Rare Dis . 1 : 46. doi : 10.1186/1750-1172-1-46 . PMC 1660532 . PMID 17109744.
^ ab Referencia, Genetics Home. «Síndrome de Bernard-Soulier». Genetics Home Reference . Archivado desde el original el 28 de julio de 2018. Consultado el 17 de julio de 2016 .
^ abc Pham A, Wang J (2007). "Síndrome de Bernard-Soulier: un trastorno plaquetario hereditario". Arch. Pathol. Lab. Med . 131 (12): 1834–6. doi :10.5858/2007-131-1834-BSAIPD. PMID 18081445. Archivado desde el original el 2022-11-11 . Consultado el 2020-02-01 .
^ abcd «Síndrome de Bernard-Soulier: aspectos prácticos, antecedentes, fisiopatología y etiología». Medscape . 22 de diciembre de 2023. Archivado desde el original el 3 de junio de 2019 . Consultado el 28 de abril de 2024 .
^ Mhawech, Paulette; Saleem, Abdus (2000). "Trastornos hereditarios de plaquetas gigantes". Revista estadounidense de patología clínica . 113 (2): 176–190. doi : 10.1309/FC4H-LM5V-VCW8-DNJU . PMID 10664620.
^ Dugdale, David. "Defectos congénitos de la función plaquetaria". NIH. Archivado desde el original el 17 de febrero de 2014. Consultado el 13 de octubre de 2012 .
^ Kanaji, T; Russell, S; Ware, J (15 de septiembre de 2002). "Mejora de la macrotrombocitopenia asociada con el síndrome de Bernard-Soulier murino". Blood . 100 (6): 2102–7. doi : 10.1182/blood-2002-03-0997 . PMID 12200373. S2CID 25130678.
^ abcde Borhany, Munira; Pahore, Zaen; ul Qadr, Zeeshan; Rehan, Mahoma; Naz, Arshi; Khan, Asif; Ansari, Saqib; Farzana, Tasneem; Nadeem, Mahoma; Raza, Syed Amir; Shamsi, Tahir (2010). "Trastornos de la coagulación en la tribu: resultado de consanguinidades en la reproducción". Revista Orphanet de Enfermedades Raras . 5 (1). doi : 10.1186/1750-1172-5-23 . ISSN 1750-1172. PMID 20822539.
^ "Síndrome de Bernard-Soulier; BSS e información sobre plaquetas gigantes. Paciente | Paciente". Paciente . 20 de abril de 2011. Archivado desde el original el 7 de febrero de 2018 . Consultado el 17 de julio de 2016 .
^ RESERVADO, INSERM US14 -- TODOS LOS DERECHOS. «Orphanet: el síndrome de Bernard Soulier». www.orpha.net . Archivado desde el original el 2017-11-16 . Consultado el 2016-07-17 .{{cite web}}: CS1 maint: nombres numéricos: lista de autores ( enlace )
^ Richmond, Caroline (10 de junio de 2006). "Jean Bernard". BMJ: British Medical Journal . 332 (7554): 1395. doi :10.1136/bmj.332.7554.1395. ISSN 0959-8138. PMC 1476743 .
Lectura adicional
Berndt, Michael C.; Andrews, Robert K. (1 de marzo de 2011). "Síndrome de Bernard-Soulier". Haematologica . 96 (3): 355–359. doi :10.3324/haematol.2010.039883. ISSN 0390-6078. PMC 3046265 . PMID 21357716.
Bick, Rodger L. (2002). Trastornos de la trombosis y la hemostasia: práctica clínica y de laboratorio. Lippincott Williams & Wilkins. ISBN 9780397516902. Recuperado el 17 de julio de 2016 .
Turgeon, Mary Louise (2005). Hematología clínica: teoría y procedimientos (4.ª ed.). Lippincott Williams & Wilkins. ISBN 9780781750073. Recuperado el 17 de julio de 2016 .
Enlaces externos
Scholia tiene un perfil de tema para el síndrome de Bernard-Soulier .