
Una reacción en cascada , también conocida como reacción dominó o reacción en tándem , es un proceso químico que comprende al menos dos reacciones consecutivas de modo que cada reacción posterior ocurre solo en virtud de la funcionalidad química formada en el paso anterior. [1] En las reacciones en cascada, no se requiere el aislamiento de intermediarios, ya que cada reacción que compone la secuencia ocurre espontáneamente. En la definición más estricta del término, las condiciones de reacción no cambian entre los pasos consecutivos de una cascada y no se agregan nuevos reactivos después del paso inicial. [1] [2] Por el contrario, los procedimientos de un solo recipiente permiten de manera similar que se lleven a cabo al menos dos reacciones consecutivas sin ningún aislamiento de intermediarios, pero no impiden la adición de nuevos reactivos o el cambio de condiciones después de la primera reacción. Por lo tanto, cualquier reacción en cascada también es un procedimiento de un solo recipiente, mientras que lo inverso no es cierto. [1] Aunque a menudo se componen únicamente de transformaciones intramoleculares, las reacciones en cascada también pueden ocurrir intermolecularmente, en cuyo caso también caen dentro de la categoría de reacciones multicomponente . [3]
Los principales beneficios de las secuencias en cascada incluyen una alta economía de átomos y una reducción de los desechos generados por los diversos procesos químicos, así como del tiempo y el trabajo necesarios para llevarlos a cabo. [1] [3] [4] La eficiencia y utilidad de una reacción en cascada se pueden medir en términos de la cantidad de enlaces formados en la secuencia general, el grado de aumento de la complejidad estructural a través del proceso y su aplicabilidad a clases más amplias de sustratos. [2] [5]
El primer ejemplo de una reacción en cascada es posiblemente la síntesis de tropinona reportada en 1917 por Robinson . [6] Desde entonces, el uso de reacciones en cascada ha proliferado en el área de la síntesis total. De manera similar, el desarrollo de la metodología orgánica impulsada por cascada también ha crecido enormemente. Este creciente interés en las secuencias en cascada se refleja en los numerosos artículos de revisión relevantes publicados en las últimas dos décadas. [1] [2] [3] [4] [5] [7] [8] [9] [10] Un área de enfoque creciente es el desarrollo de la catálisis asimétrica de los procesos en cascada mediante el empleo de organocatalizadores quirales o complejos de metales de transición quirales. [3] [7] [10] [11]
La clasificación de las reacciones en cascada a veces es difícil debido a la naturaleza diversa de los muchos pasos de la transformación. KC Nicolaou etiqueta las cascadas como nucleófilas/electrófilas, radicales, pericíclicas o catalizadas por metales de transición, según el mecanismo de los pasos involucrados. En los casos en los que se incluyen dos o más clases de reacción en una cascada, la distinción se vuelve bastante arbitraria y el proceso se etiqueta de acuerdo con lo que podría considerarse el "tema principal". [4] Para destacar la notable utilidad sintética de las reacciones en cascada, la mayoría de los ejemplos a continuación provienen de las síntesis totales de moléculas complejas.
Las cascadas nucleofílicas/electrofílicas se definen como las secuencias en cascada en las que el paso clave constituye un ataque nucleofílico o electrofílico. [4]
Un ejemplo de tal cascada se ve en la síntesis enantioselectiva corta del antibiótico de amplio espectro (–)-cloranfenicol, reportada por Rao et al. (Esquema 1). [3] [12] En este caso, el epoxi-alcohol quiral 1 se trató primero con dicloroacetonitrilo en presencia de NaH. El intermedio resultante 2 luego experimentó una reacción en cascada mediada por BF 3 ·Et 2 O. La apertura intramolecular del anillo de epóxido produjo el intermedio 3 , que, después de una hidrólisis in situ facilitada por un exceso de BF 3 ·Et 2 O, proporcionó (–)-cloranfenicol ( 4 ) con un rendimiento general del 71%. [3] [12]

También se empleó una cascada nucleofílica en la síntesis total del producto natural pentaleneno (Esquema 2). [4] [13] En este procedimiento, el éster escuarato 5 se trató con (5-metilciclopent-1-en-1-il)litio y propinillitio . Los dos ataques nucleofílicos ocurrieron predominantemente con adición trans para producir el intermedio 6 , que espontáneamente experimentó una apertura electrocíclica 4π-conrotatoria del anillo de ciclobuteno. La especie conjugada resultante 7 se equilibró con el confórmero 8 , que experimentó más fácilmente una electrociclación 8π-conrotatoria al intermedio altamente deformado 9. El potencial para liberar la deformación dirigió la protonación de 9 de modo que la especie 10 se obtuvo selectivamente. La cascada se completó con una condensación aldólica intramolecular que produjo el producto 11 con un rendimiento general del 76%. Una elaboración posterior proporcionó el objetivo (±)-pentaleno ( 12 ). [4] [13]

Una subcategoría de secuencias nucleofílicas/electrofílicas está constituida por cascadas organocatalíticas, en las que el ataque nucleofílico clave es impulsado por la organocatálisis.
Se empleó una cascada organocatalítica en la síntesis total del producto natural harzifilona, informado por Sorensen et al. en 2004 (Esquema 3). [4] [14] En este caso, el tratamiento del material de partida enona 13 con el organocatalizador 14 produjo el intermedio 15 mediante adición conjugada. La ciclización posterior mediante la adición intramolecular de Michael del enolato en el triple enlace del sistema produjo la especie 16 , que produjo el intermedio 17 después de la transferencia de protones y la tautomerización. La cascada se completó mediante la eliminación del organocatalizador y un cierre espontáneo del anillo 6π-electrocíclico de la cis -dienona resultante 18 a (+)-harzifilona ( 19 ) con un rendimiento general del 70%. [4] [14]

En 2006, Raabe et al. informaron sobre una cascada organocatalítica triple excepcional. Los aldehídos lineales ( 20 ), los nitroalquenos ( 21 ) y los aldehídos α , β -insaturados ( 22 ) podrían condensarse juntos de forma organocatalítica para producir carbaldehídos de ciclohexano tetra -sustituidos ( 24 ) con una diastereoselectividad moderada a excelente y un control enantiométrico completo (Esquema 4). La transformación está mediada por el organocatalizador derivado de prolina, fácilmente disponible, 23. [ 15]

Se propuso que la transformación procediera a través de una secuencia de adición de Michael/adición de Michael/condensación aldólica (Esquema 5). [15] En el primer paso, la adición de Michael del aldehído 20 al nitroalqueno 21 ocurre a través de catálisis de enamina, produciendo nitroalcano 25. La condensación del aldehído α , β -insaturado 22 con el organocatalizador facilita luego la adición conjugada de 25 para dar la enamina intermedia 26 , que es propensa a sufrir una condensación aldólica intramolecular a la especie de iminio 27. El organocatalizador 23 se regenera por hidrólisis, junto con el producto 24 , cerrando así el ciclo de triple cascada. [15]

Las cascadas de radicales son aquellas en las que el paso clave constituye una reacción radicalaria. La alta reactividad de las especies de radicales libres hace que los enfoques sintéticos basados en radicales sean decididamente adecuados para las reacciones en cascada. [4]
Uno de los ejemplos más ampliamente reconocidos de la utilidad sintética de las cascadas de radicales es la secuencia de ciclización empleada en la síntesis total de (±)-hirsuteno, en 1985 (Esquema 6). [4] [16] En este caso, el yoduro de alquilo 28 se convirtió en el intermediario radical primario 29 , que experimentó una ciclización 5- exo -trig para producir la especie reactiva 30. Una ciclización radical 5- exo -dig posterior condujo al intermediario 31 , que al extinguirse dio el (±)-hirsuteno objetivo ( 32 ) con un rendimiento general del 80%. [4] [16]

También se utilizó un proceso radical en cascada en una de las síntesis totales de (–)-morfina (Esquema 7). [4] [17] [18] El bromuro de arilo 33 se convirtió en la especie radical correspondiente 34 mediante tratamiento con hidruro de tri- n -butilestaño. Luego se produjo una ciclización 5- exo -trig para dar el intermedio 35 de forma estereoselectiva en virtud de la estereoquímica del enlace éter. En el siguiente paso de la cascada, las restricciones geométricas de 35 prohíben la vía de ciclización 5- exo -trig cinéticamente favorecida ; en su lugar, se obtuvo la especie radical bencílico secundaria 36 mediante una ciclización 6- endo -trig geométricamente permitida. La eliminación posterior del radical fenil sulfinilo proporcionó el producto 37 con un rendimiento general del 30 %, que se elaboró posteriormente para (–)-morfina ( 38 ). [4] [17] [18]

Posiblemente el tipo de proceso más ampliamente encontrado en las transformaciones en cascada, las reacciones pericíclicas incluyen cicloadiciones, reacciones electrocíclicas y reordenamientos sigmatrópicos. [4] Aunque algunos de los casos mencionados anteriormente de cascadas nucleofílicas/electrófilas y radicales involucraron procesos pericíclicos, esta sección contiene solo secuencias en cascada que están compuestas únicamente de reacciones pericíclicas o en las que dicha reacción posiblemente constituye el paso clave.
Un ejemplo representativo de una cascada pericíclica es la cascada del ácido endiándrico descrita por Nicolaou et al. en 1982 (Esquema 8). [4] [19] En este caso, el sistema altamente insaturado 39 se hidrogenó primero a la especie de tetraeno conjugado 40 , que al calentarse experimentó un cierre de anillo electrocíclico 8π-conrotatorio, produciendo el intermedio cíclico 41. Una segunda electrociclación espontánea, esta vez un cierre de anillo 6π-disrotatorio, convirtió a 41 en la especie bicíclica 42 , cuya geometría y estereoquímica favorecieron una reacción de Diels-Alder intramolecular posterior. El éster metílico del ácido endiándrico B ( 43 ) se obtuvo así con un rendimiento general del 23%. [4] [19]

Se empleó una secuencia pericíclica que involucra reacciones de heterocicloadición intramolecular en la síntesis total del alcaloide natural (–)-vindorosina (Esquema 9). [4] [20] Se logró un acceso rápido al objetivo a partir de una solución de 1,3,4-oxadiazol 44 en triisopropilbenceno sometido a altas temperaturas y presión reducida. Primero se produjo una reacción hetero-Diels-Alder de demanda inversa de electrones para dar el intermedio 45. La pérdida de nitrógeno termodinámicamente favorable generó la especie que contiene 1,3-dipolo 46. Una cicloadición intramolecular espontánea [3+2] del 1,3-dipolo y el sistema indólico formó luego el endoproducto 47 con un rendimiento total del 78% . Una elaboración posterior produjo el producto natural objetivo 48. [4] [20]

La síntesis total de (–)-colombiasina A reportada en 2005 por el grupo Harrowven incluyó una cascada electrocíclica (Esquema 10). [4] [21] Cuando se sometió a calor a través de irradiación de microondas, el derivado de escuadrato 49 experimentó una apertura electrocíclica del anillo de ciclobuteno, seguida de un cierre de anillo 6π-electrocíclico que produjo el intermedio bicíclico 51. Su tautomerización dio la especie aromática 52 , que al exponerse al aire se oxidó al producto 53 con un rendimiento general del 80%. La (–)-colombiasina A objetivo ( 54 ) se obtuvo luego a partir de 53 a través de una reacción de Diels-Alder facilitada por calor seguida de la escisión del grupo protector terc -butilo. [4] [21]

Ciertos [2,2]paraciclofanos también pueden obtenerse mediante cascadas pericíclicas, como informó el grupo Hopf en 1981 (Esquema 11). [1] [22] En esta secuencia, una reacción de Diels-Alder entre 1,2,4,5-hexatetraeno 55 y dienófilo 56 formó primero el intermedio altamente reactivo 57 , que posteriormente se dimerizó para producir [2,2]paraciclofano 58. [1] [22 ]
![Esquema 11. Secuencia pericíclica para la síntesis de [2,2]paraciclofanos](http://upload.wikimedia.org/wikipedia/commons/thumb/b/be/Scheme_11_-_peri_-_pcyclophane.svg/1280px-Scheme_11_-_peri_-_pcyclophane.svg.png)
Las secuencias en cascada catalizadas por metales de transición combinan la novedad y el poder de la química organometálica con la utilidad sintética y la economía de las reacciones en cascada, proporcionando un enfoque aún más deseable desde el punto de vista ecológico y económico para la síntesis orgánica. [4]
Por ejemplo, se utilizó catálisis de rodio para convertir monoterpenos acíclicos del tipo 59 en productos de 4 H -cromeno en una cascada de hidroformilación (Esquema 12). [8] [23] Primero, la hidroformilación selectiva catalizada por rodio del enlace de olefina menos impedido estéricamente en 59 produjo el aldehído insaturado 60 , que bajo las mismas condiciones luego se convirtió en el intermedio 61 a través de una reacción de carbonilo-eno. Una segunda hidroformilación catalizada por rodio a la especie 62 fue seguida por condensación para formar productos de 4 H -cromeno del tipo 63 en un rendimiento general del 40%. [8] [23]

La catálisis de rodio también se empleó para iniciar una cascada de ciclización/cicloadición en la síntesis de un tigliano reportada por el grupo Dauben (Esquema 13). [2] [24] El tratamiento de diazoimida 64 con dímero de acetato de rodio (II) generó un carbenoide que produjo el iluro reactivo 65 después de una ciclización intramolecular con el grupo carbonilo vecino. Luego se produjo espontáneamente una cicloadición intramolecular [3+2] para producir el tigliano objetivo 66. [2] [24]

La cicloadición intramolecular formal [4+2] de 1,6-eninos del tipo 67 mediada por catálisis de oro es otro ejemplo de una cascada catalizada por metales de transición (Esquema 14). [25] [26] Una variedad de 1,6-eninos reaccionaron en condiciones suaves en presencia de complejos de Au(I) 68a – b para producir los productos tricíclicos 69 en rendimientos moderados a excelentes. [25] [26]
![Esquema 14. Cicloadición intramolecular formal [4+2] catalizada por oro de 1,6-eninos](http://upload.wikimedia.org/wikipedia/commons/thumb/2/2b/Scheme_14_-_metal_-_gold_enyne.svg/1280px-Scheme_14_-_metal_-_gold_enyne.svg.png)
Se propuso que esta cicloadición formal procediera a través del proceso en cascada que se muestra en el Esquema 15. [25] [26] La complexación del 1,6-enino 67 con la forma catiónica del catalizador produce el intermedio 70 , en el que el triple enlace activado es atacado por la funcionalidad de olefina para producir ciclopropano sustituido 71. La apertura electrofílica del anillo de tres miembros forma la especie catiónica 72 , que experimenta una reacción de tipo Friedel Crafts y luego se rearomatiza para dar el producto tricíclico 69. [25] [26] Debido a la naturaleza de la interacción de los complejos de oro con sistemas insaturados, este proceso también podría considerarse una cascada electrofílica.
![Esquema 15. Proceso en cascada propuesto en la cicloadición intramolecular formal [4+2] de 1,6-eninos](http://upload.wikimedia.org/wikipedia/commons/thumb/b/ba/Scheme_15_-_metal_-_gold_enyne_mech.svg/1280px-Scheme_15_-_metal_-_gold_enyne_mech.svg.png)
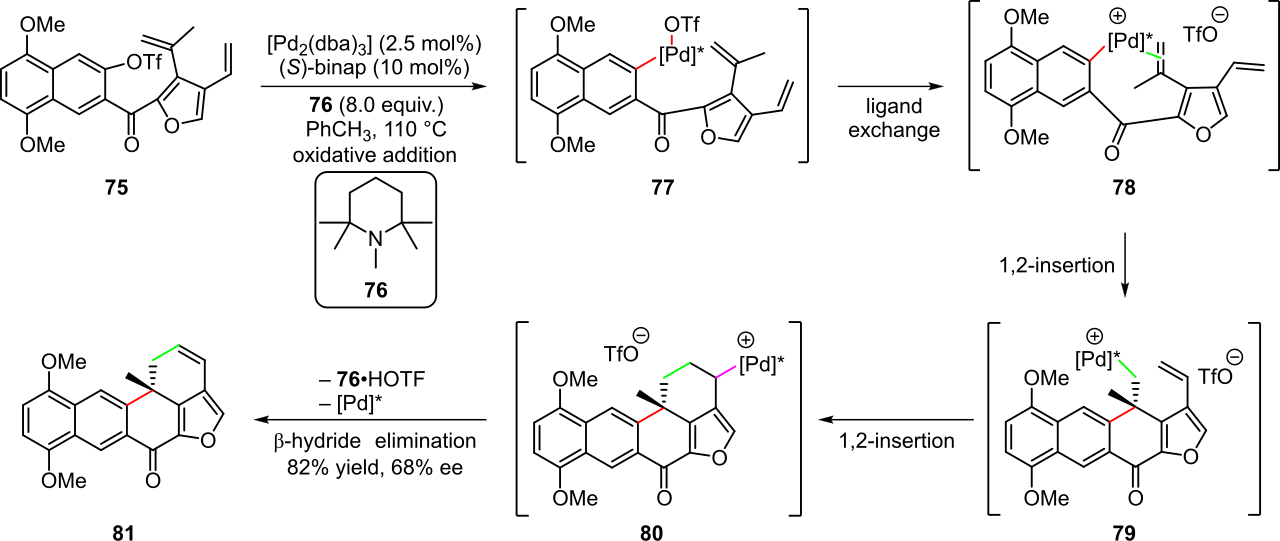
Un ejemplo de cascadas catalizadas por paladio está representado por la ciclización asimétrica de Heck de polieno utilizada en la preparación de (+)-xestoquinona a partir del sustrato triflato 75 (Esquema 16). [4] [27] La adición oxidativa del enlace arilo-triflato en el complejo de paladio(0) en presencia del ligando de difosfina quiral ( S )-binap produce el complejo de paladio(II) quiral 77 . Este paso es seguido por la disociación del anión triflato, la asociación de la olefina vecina y la 1,2-inserción del grupo naftilo en la olefina para producir el intermedio 79 . Luego ocurre una segunda inserción migratoria en el grupo olefina restante seguida de una β -eliminación para producir el producto 81 con un rendimiento general del 82 % y con una enantioselectividad moderada. El catalizador de paladio(0) también se regenera en este paso, lo que permite reiniciar la cascada. [4] [27]
Las reacciones en tándem de varios pasos (o reacciones en cascada) son una secuencia de transformaciones químicas (generalmente más de dos pasos) que ocurren consecutivamente para convertir un material de partida en un producto complejo. [28] Este tipo de reacciones orgánicas están diseñadas para construir estructuras difíciles que se encuentran en la síntesis total de productos naturales .
En la síntesis total del antibiótico ionóforo espirocetal rutiennocina 1 (Fig. 1), el esqueleto espirocetal central se construyó mediante una reacción en tándem de varios pasos (Fig. 2). [29] El fragmento A y el fragmento B se acoplaron en un solo paso para formar el intermediario clave G que podría elaborarse aún más para proporcionar el producto final rutiennocina.
En esta reacción en tándem se produjeron cuatro transformaciones químicas. En primer lugar, el tratamiento del fragmento A con n-butillitio formó un anión de carbono que atacó la parte de yoduro de alquilo del fragmento B para generar el intermedio C (paso 1). A continuación, se formó un derivado de 3,4-dihidropirano D mediante una reacción de eliminación mediada por una base en el intermedio C (paso 2). El grupo protector de la fracción 1,3- diol del intermedio D se eliminó mediante un tratamiento ácido para dar el producto diol E (paso 3). El producto espirocetal G se generó mediante una reacción de formación de cetal intramolecular . Esta reacción en tándem de varios pasos simplificó en gran medida la construcción de esta compleja estructura espirocetal y facilitó el camino hacia la síntesis total de rutiennocina.
