La miopatía nemalínica (también llamada miopatía en bastones o miopatía en bastones nemalínicos ) es un trastorno neuromuscular congénito , a menudo hereditario , con muchos síntomas que pueden presentarse, como debilidad muscular, hipoventilación , disfunción de la deglución y deterioro de la capacidad del habla. La gravedad de estos síntomas varía y puede cambiar a lo largo de la vida hasta cierto punto. La prevalencia se estima en 1 de cada 50.000 nacidos vivos. [1] Es la miopatía no distrófica más común. [2] [3]
"Miopatía" significa enfermedad muscular. Las fibras musculares de una persona con miopatía nemalínica contienen varillas con forma de filamento [4] , a veces llamadas cuerpos nemalínicos. [5] Si bien las varillas son diagnósticas del trastorno, es más probable que sean un subproducto del proceso de la enfermedad en lugar de causar una disfunción por sí mismas. Las personas con miopatía nemalínica (NM) generalmente experimentan un desarrollo motor retrasado, o ningún desarrollo motor en casos graves, y puede ocurrir debilidad en todos los músculos esqueléticos, como los músculos de los brazos, las piernas, el torso, los flexores del cuello, la garganta y la cara. La debilidad tiende a ser más grave en los músculos proximales que en los músculos distales. Los músculos oculares normalmente están intactos.
El trastorno se suele clasificar clínicamente en grupos con amplios rangos de gravedad superpuesta, desde la forma neonatal más grave que es incompatible con la vida , hasta una forma tan leve que puede no diagnosticarse ya que la persona parece funcionar en el extremo más bajo de la fuerza normal y la adecuación respiratoria. La miopatía nemalínica de aparición tardía esporádica (SLONM) no es un trastorno congénito y se considera una enfermedad muscular diferente de la NM, que tiene su inicio en el nacimiento o la primera infancia. [6] Los problemas respiratorios suelen ser una preocupación principal para las personas con todas las formas de NM, y las infecciones respiratorias son bastante comunes. La NM acorta la esperanza de vida, particularmente en las formas más graves, pero la atención agresiva y proactiva permite a la mayoría de las personas sobrevivir e incluso llevar una vida activa.
La miopatía nemalínica es una de las enfermedades neuromusculares cubiertas por la Asociación de Distrofia Muscular en los Estados Unidos.
Los síntomas varían de persona a persona. Los niños pequeños y los bebés carecen de movimiento y tienen dificultad para comer y respirar. En los niños pequeños que no reciben un diagnóstico inmediato al nacer, estos suelen ser los primeros síntomas visibles. Uno de ellos es la hinchazón de la cara en zonas desproporcionadas. Otros ejemplos en los recién nacidos son el balanceo y la dificultad para moverse. Otros síntomas incluyen debilidad muscular en el cuello y la zona superior de la caja torácica. En los adultos, el síntoma más común son los problemas respiratorios. [7] Otros síntomas en los adultos pueden variar desde impedimentos del habla leves a graves. Es común que se diagnostique escoliosis en relación con la miopatía nemalínica. [8] A medida que los bebés que tienen NM se desarrollan y alcanzan la edad en la que deberían empezar a caminar, muchos tardan más de lo normal debido a la falta de músculo o simplemente a la fatiga muscular. [9]
Dado que los músculos faciales están involucrados en la toma de control de la NM, a menudo se observan caras alargadas y una mandíbula inferior en las personas con NM. Las personas afectadas por NM generalmente comenzarán a sentir agotamiento muscular entre los 20 y los 50 años. La NM no suele ser progresiva. El reflujo gastroesofágico , aunque no es común, está asociado con la NM. Pueden ocurrir anomalías cardíacas como resultado de la NM, pero la probabilidad de que eso suceda no es alta. [7]
La mayoría de los niños con NM leve acaban caminando de forma independiente, aunque a menudo a una edad más avanzada que sus compañeros. Algunos utilizan sillas de ruedas u otros dispositivos, como andadores o aparatos ortopédicos, para mejorar su movilidad. Las personas con NM grave suelen tener un movimiento limitado de las extremidades y utilizan sillas de ruedas a tiempo completo. Debido a la debilidad de los músculos del tronco, las personas con NM son propensas a la escoliosis , que suele desarrollarse en la infancia y empeora durante la pubertad. Muchas personas con NM se someten a una cirugía de fusión espinal para enderezar y estabilizar la espalda. [ cita requerida ]
Aunque los pacientes con miopatía nemalínica suelen tener una movilidad en las articulaciones que supera los límites normales, a medida que envejecen, suelen aparecer deformidades articulares y escoliosis . [10] Si la persona con miopatía nemalínica vigila sus articulaciones desde el principio, los problemas con ellas se pueden detectar cuando aparecen y su progresión se puede retrasar. El tratamiento de los problemas articulares varía desde ejercicios de estiramiento con fisioterapia hasta la introducción quirúrgica de aparatos ortopédicos. Los beneficios del ejercicio en personas con miopatía nemalínica aún se están estudiando, sin embargo, los investigadores han visto mejoras en la función muscular con el ejercicio de baja intensidad. Se debe evitar el ejercicio vigoroso y el uso de pesas pesadas. [ cita requerida ]
La atención a los problemas respiratorios es fundamental para la salud de muchas personas con NM. Los bebés con NM grave con frecuencia experimentan dificultad respiratoria al nacer o poco después, aunque esto solo se encuentra en las formas más raras. Aunque el compromiso respiratorio puede no ser evidente de inmediato en personas con NM intermedia o leve, a menudo, pero no siempre, existe en cierta medida. Como en muchos trastornos neuromusculares, la hipoventilación puede comenzar de manera insidiosa y puede causar problemas de salud graves si no se remedia con el uso de dispositivos mecánicos no invasivos para ayudar a respirar, en particular durante la noche. [ cita requerida ]
La debilidad de los músculos bulbares (de la garganta) es una característica principal de la miopatía nemalínica. Las personas con las formas más graves de NM no pueden tragar y reciben su nutrición a través de sondas de alimentación . La mayoría de las personas con NM intermedia y leve toman parte o toda su nutrición por vía oral. El deterioro de los músculos bulbares también puede provocar dificultades para comunicarse. Las personas con NM a menudo tienen habla hipernasal como resultado del cierre deficiente del puerto velofaríngeo (entre el paladar blando y la parte posterior de la garganta). Esto puede corregirse quirúrgicamente. Las habilidades comunicativas pueden mejorarse mediante terapia del habla, dispositivos protésicos orales, cirugía y dispositivos de comunicación aumentativa . La NM no tiene ningún impacto en la cognición o la inteligencia. [ cita requerida ]
La expresión física de la miopatía nemalínica varía mucho, pero la debilidad suele concentrarse en los músculos proximales, en particular los músculos respiratorios, bulbares y del tronco. [11] Las personas con NM grave muestran síntomas evidentes al nacer, mientras que las que tienen NM intermedia o leve pueden parecer inicialmente no afectadas. Con frecuencia se observa que los bebés con NM están "flojos" e hipotónicos . Los niños que nacen con NM suelen ganar fuerza a medida que crecen, aunque el efecto de la debilidad muscular en las características corporales puede hacerse más evidente con el tiempo. Los adultos con NM suelen tener un físico muy delgado. [11]
La miopatía nemalínica es causada por mutaciones en uno de al menos 11 genes diferentes. [2] [12] La miopatía nemalínica es un trastorno clínicamente y genéticamente heterogéneo y pueden presentarse formas autosómicas dominantes y autosómicas recesivas . El diagnóstico se realiza en base a signos clínicos como debilidad muscular, reflejos osteotendinosos ausentes o bajos (hiporeflexia) y paladar muy arqueado, junto con agregados densos en electrones, llamados bastones nemalínicos, que se observan a nivel microscópico dentro de las fibras musculares. La confirmación genética a través de la identificación de una mutación genética conocida en el paciente también es un componente importante del diagnóstico. [13] [14]
Las dos mutaciones genéticas más comunes que causan miopatía nemalínica se encuentran en NEB o ACTA1 . [15] Las mutaciones del gen NEB generalmente resultan en síntomas presentes al nacer o que comienzan en la primera infancia. Esta mutación resulta en aproximadamente el 50% de los pacientes afectados de miopatía nemalínica. La vía de herencia más común para aquellos con mutaciones en NEB es autosómica recesiva en la que cada padre lleva una copia mutada junto con una copia de funcionamiento normal del gen, y pasan la copia mutada a su descendencia. En algunos casos, ocasionalmente con mutaciones ACTA1 , NM puede ser causada por un patrón de herencia de dominancia autosómica. Esta mutación resulta en aproximadamente el 15 al 25 por ciento de los casos de NM. [16] Una razón por la que esto es menor es porque NM está asociada con mutaciones de novo en ACTA1 , que ocurren espontáneamente en el óvulo o el espermatozoide. [9] Cuando la afección es hereditaria, cada embarazo con las mismas parejas tiene el mismo riesgo de transmitir los genes mutados a la descendencia. También pueden ocurrir nuevas mutaciones (de novo) que causen NM y se ha encontrado que las mutaciones de novo ocurren con mayor frecuencia en el gen ACTA1 . [16] MYPN es el último gen encontrado relacionado con NM. El riesgo de todos los casos de miopatía nemalínica es el mismo en hombres y mujeres. [9]

Las capacidades físicas de una persona determinada con NM no se correlacionan bien ni con el genotipo ni con la patología muscular observada en la biopsia. [18]
Las células musculares se contraen mediante complejos procesos mecánicos y químicos. Si se altera alguna parte del proceso o la estructura, es probable que se produzca una disfunción, como en el caso de las personas con variaciones genéticas. En las personas con miopatía nemalínica, la contracción muscular se ve afectada negativamente. A nivel de microscopio electrónico, a menudo se pueden ver componentes en forma de varilla en algunas de las células musculares y, cuando se ven, son diagnósticos de la afección denominada miopatía por varillas nemalínicas. La presencia de estas varillas no causa debilidad muscular en sí misma, sino que aparecen como resultado de algo que va mal dentro de la fibra muscular. No existe ninguna conexión entre la cantidad de varillas que se encuentran en las células musculares y el grado de debilidad que tiene una persona. Todas las diferentes mutaciones genéticas que conducen a la afección denominada miopatía nemalínica que se han encontrado hasta ahora se encuentran en genes que codifican diferentes componentes del sarcómero. [11] En las células musculares normales, las diversas partes de las fibras musculares que forman el sarcómero se distribuyen uniformemente en un patrón para una contracción muscular eficaz. La evidencia sugiere que algunos tipos de NM afectan la disposición de estas fibras musculares, provocando que los músculos no puedan contraerse de manera tan eficiente o efectiva. [ cita requerida ]

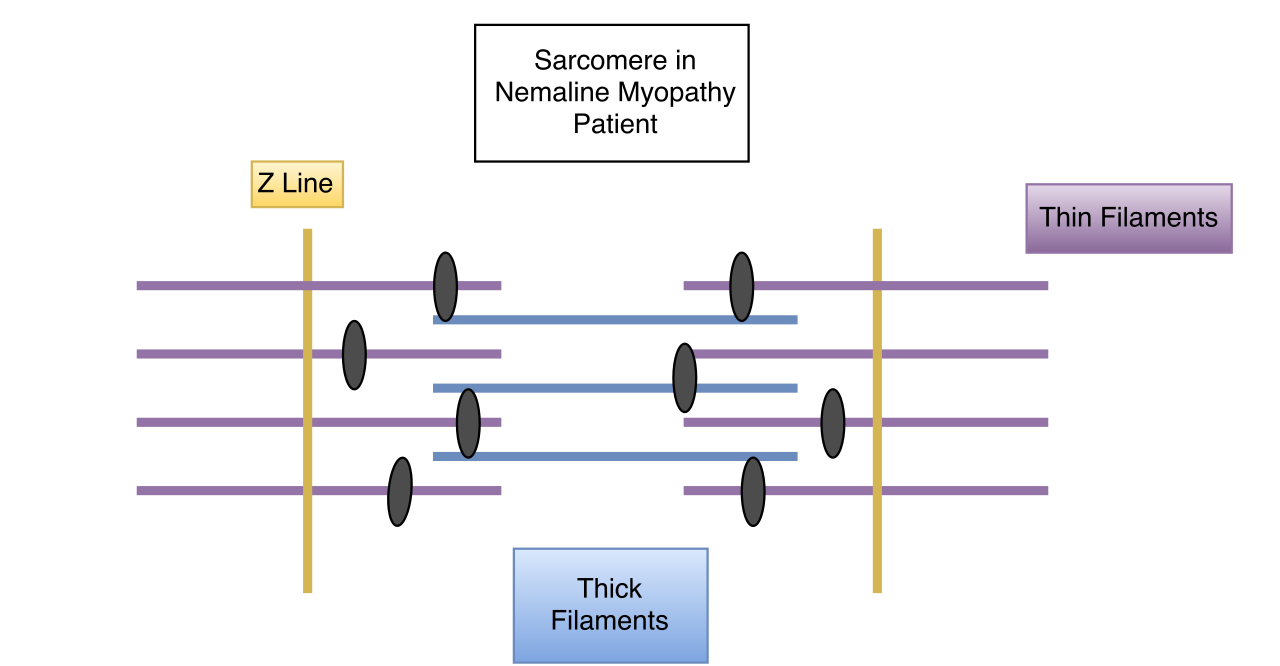
La miopatía nemalínica suele ser genética y muestra rasgos en el individuo afectado desde el nacimiento o a una edad temprana. Sin embargo, hay algunos casos en los que los síntomas de la miopatía nemalínica no aparecen hasta la edad adulta. Estos casos no suelen ser genéticos. De los genes que se han relacionado con la miopatía nemalínica, la mayoría también están involucrados en la codificación de proteínas en los sarcómeros de las células musculares. [11] Los músculos respiratorios suelen verse más afectados que otros grupos de músculos esqueléticos. El músculo cardíaco no suele verse afectado en la miopatía nemalínica; sin embargo, en los casos en que sí lo hace, los pacientes suelen presentar miocardiopatía dilatada. Los músculos oculares suelen estar intactos. [ cita requerida ]
Los diferentes genes cuyas mutaciones dan lugar a los distintos tipos de miopatías nemalínicas afectan a las células y al cuerpo de la persona de forma diferente. El primer tipo de miopatía nemalínica identificado se debe al gen de la α-tropomiosina lenta TPM3 y varía de un caso a otro en cuanto a su gravedad. En este tipo de miopatía nemalínica, las personas afectadas son más débiles y tienen más afectadas las extremidades inferiores que las superiores. [11]
Como se mencionó anteriormente, la forma genética más común de NM es causada por una mutación en el gen de la nebulina, llamado Nebulin, [19] [20] y tiene un rango de niveles de gravedad. Todos los casos publicados hasta el momento en los que se cree que la NM es causada por una mutación en el gen NEB han sido autosómicos recesivos y son la causa más común de miopatía nemalínica. [21] Los pacientes con este tipo de NM se ven más afectados en los músculos de la cabeza, en lugar de los músculos proximales en el centro del cuerpo. En consecuencia, los pacientes con esta mutación genética a menudo no pueden levantar la cabeza y hablar con una voz nasal. Ha habido casos que sugieren que este tipo de NM puede hacer que los pacientes tengan un intelecto más alto. [11]
Un tercer tipo de miopatía nemalínica en el gen de la α-actina del músculo esquelético ACTA1 se debe a una mutación nula recesiva. [11] Estos pacientes no siempre muestran los cuerpos nemalínicos típicos en sus células musculares. La única anomalía que presentan es una distribución anormal de las fibras musculares.
Existen otros tipos de mutaciones identificadas que provocan miopatías nemalinas. Una afecta a los músculos esqueléticos lentos y otra conduce a la formación de cuerpos nemalinos y otras estructuras anormales similares a núcleos en los músculos del paciente. [ cita requerida ]
En la actualidad, la miopatía nemalínica no tiene cura. La miopatía nemalínica es una enfermedad muy rara que solo afecta a 1 de cada 50.000 de media, aunque estudios recientes muestran que este número es incluso menor. Existen diversos tratamientos para minimizar los síntomas de la enfermedad. Los tratamientos y procedimientos para ayudar a los pacientes con miopatía nemalínica varían en función de la gravedad de la enfermedad. Una posible adaptación podría ser el uso de un estabilizador, como un corsé. Otros medios incluyen estiramientos moderados y ejercicio moderado para ayudar a los músculos objetivo a mantener la máxima salud. [22] A medida que las personas con NM crecen y se desarrollan a lo largo de sus vidas, es importante que acudan a una variedad de profesionales de la salud con regularidad, incluidos un neurólogo , un fisioterapeuta y otros, como logopedas y psicólogos , para ayudar tanto al paciente como a la familia a adaptarse a la vida cotidiana. [10]
Aunque no existe cura para la NM, es posible y común que muchas personas lleven una vida activa y saludable incluso en casos moderados a severos. [23] La investigación continúa buscando formas de mejorar los síntomas debilitantes y alargar la vida de manera cualificada para los afectados. Algunas personas han experimentado mejoras leves en el manejo de las secreciones, el nivel de energía y el funcionamiento físico con suplementos de L-tirosina, un aminoácido que está disponible en los centros de salud. [24] Algunos síntomas pueden empeorar a medida que el paciente envejece. La pérdida de masa muscular aumenta con la edad de forma natural, pero es aún más significativa en el caso de la miopatía nemalínica. [25]
Se han puesto a disposición de la comunidad de NM nuevos recursos de investigación, como el CMDIR (registro) y el CMD-TR (biorepositorio). Estos dos recursos conectan a las familias y a las personas interesadas en participar en investigaciones con los científicos que tienen como objetivo tratar o curar la NM. Algunas investigaciones sobre la NM buscan comprender mejor los efectos moleculares que tienen las mutaciones genéticas en las células musculares y en el resto del cuerpo [26] y observar cualquier conexión que la NM pueda tener con otras enfermedades y complicaciones de salud.
La "miopatía en bastones" fue identificada por primera vez por Douglas Reye , un médico australiano, en 1958. [27] Sin embargo, los resultados de Reye nunca se publicaron porque otro médico descartó su hallazgo de bastones en el tejido muscular como un artefacto de la biopsia. Cuarenta años después, se confirmó que el paciente de Reye con "miopatía en bastones" tenía miopatía nemalínica. Desde entonces, otro grupo de investigadores australianos ha publicado un artículo en reconocimiento a Reye por su trabajo. [28]
La "miopatía nemalínica" fue nombrada por primera vez en un artículo publicado en 1963 por los investigadores norteamericanos PE Cohen y GM Shy. Shy y su equipo descubrieron estructuras con forma de varilla en las fibras musculares de pacientes con debilidad muscular al realizar biopsias musculares en múltiples pacientes. [22] Los laboratorios que realizan investigaciones sobre la NM se encuentran en todo el mundo, especialmente en Estados Unidos, Canadá, Inglaterra, Finlandia y Australia.
En 1999, David McDougall lanzó la Comunidad NM [29] y el primer sitio web (www.nemaline.org [30] ) para la miopatía nemalínica, y en octubre de 2004, el Grupo de Apoyo a la Miopatía Nemalínica organizó la primera Conferencia sobre Miopatía Nemalínica en Toronto, Canadá. Desde entonces se han celebrado muchas más conferencias y eventos sociales, y todos los eventos organizados desde 2008 han sido copatrocinados por A Foundation Building Strength for Nemaline Myopathy (AFBS), [31] la única fundación centrada en apoyar el desarrollo de tratamientos y eventos sociales para la comunidad NM. En marzo de 2006, Niki Shisler publicó un libro, Fragile , en el que relataba sus experiencias en torno al nacimiento de dos hijos gemelos con NM grave. En 2014, un equipo de expertos colaboró con personas afectadas y familias que cuidaban a alguien con una miopatía congénita para desarrollar la primera guía sobre cómo manejar la vida con una miopatía congénita [ cita requerida ]