La enfermedad de células falciformes ( ECF ), también llamada simplemente anemia falciforme , es un grupo de trastornos sanguíneos relacionados con la hemoglobina que generalmente se heredan . [2] El tipo más común se conoce como anemia de células falciformes . [2] Resulta en una anomalía en la hemoglobina, una proteína transportadora de oxígeno que se encuentra en los glóbulos rojos . [2] Esto conduce a una forma rígida, similar a una hoz, en determinadas circunstancias. [2] Los problemas de la enfermedad de células falciformes suelen comenzar alrededor de los 5 a 6 meses de edad. [1] Pueden desarrollarse varios problemas de salud, como ataques de dolor (conocidos como crisis de células falciformes ) en las articulaciones, anemia , hinchazón en las manos y los pies, infecciones bacterianas , mareos [9] y accidente cerebrovascular . [1] El dolor a largo plazo puede desarrollarse a medida que las personas envejecen. [2] La esperanza de vida promedio en el mundo desarrollado es de 40 a 60 años. A menudo empeora con la edad. [10] Todos los órganos principales se ven afectados por la enfermedad de células falciformes. El hígado, el corazón, los riñones, la vesícula biliar, los ojos, los huesos y las articulaciones también pueden sufrir daños debido a las funciones anormales de las células falciformes y su incapacidad para fluir correctamente a través de los vasos sanguíneos pequeños. [11]
La enfermedad de células falciformes se produce cuando una persona hereda dos copias anormales del gen de la β-globina ( HBB ) que produce hemoglobina, una de cada progenitor. [3] Este gen se encuentra en el cromosoma 11. [12] Existen varios subtipos, dependiendo de la mutación exacta en cada gen de hemoglobina. [2] Un ataque puede ser desencadenado por cambios de temperatura, estrés, deshidratación y gran altitud. [1] Una persona con una sola copia anormal no suele tener síntomas y se dice que tiene el rasgo de células falciformes . [3] A estas personas también se las conoce como portadoras . [5] El diagnóstico se realiza mediante un análisis de sangre , y algunos países realizan pruebas a todos los bebés al nacer para detectar la enfermedad. [4] El diagnóstico también es posible durante el embarazo. [4]
El cuidado de las personas con enfermedad de células falciformes puede incluir la prevención de infecciones con vacunación y antibióticos , una ingesta elevada de líquidos, suplementos de ácido fólico y analgésicos . [5] [6] Otras medidas pueden incluir la transfusión de sangre y el medicamento hidroxicarbamida (hidroxiurea). [6] En 2023, se aprobaron nuevas terapias genéticas . [13] [14] Un pequeño porcentaje de personas puede curarse mediante un trasplante de células de médula ósea .
En 2015 [update], aproximadamente 4,4 millones de personas padecen anemia de células falciformes, mientras que otros 43 millones tienen el rasgo de células falciformes. [7] [15] Se cree que aproximadamente el 80% de los casos de anemia de células falciformes ocurren en África subsahariana . [16] También ocurre en menor grado en partes de la India , el sur de Europa , Asia occidental , el norte de África y entre personas de origen africano (subsaharianos) que viven en otras partes del mundo. [17] En 2015, resultó en aproximadamente 114.800 muertes. [8] La afección fue descrita por primera vez en la literatura médica por el médico estadounidense James B. Herrick en 1910. [18] [19] En 1949, EA Beet y JV Neel determinaron su transmisión genética. [19] En 1954, se describió el efecto protector contra la malaria del rasgo de células falciformes. [19]
_(14768428942).jpg/1280px-The_diseases_of_infancy_and_childhood_(1920)_(14768428942).jpg)


Los síntomas de la enfermedad de células falciformes suelen aparecer en la primera infancia. La gravedad de los síntomas puede variar de una persona a otra, al igual que la frecuencia de los episodios de crisis. [20] [16] La enfermedad de células falciformes puede provocar diversas complicaciones agudas y crónicas , varias de las cuales tienen una alta tasa de mortalidad. [21]
Las características habituales del primer episodio que lleva al diagnóstico de anemia falciforme dependen de la edad del niño. Cuando la enfermedad se presenta durante el primer año de vida, el problema más común es un episodio de dolor e hinchazón en las manos y los pies, conocido como dactilitis o “síndrome mano-pie”. La palidez, la ictericia y la fatiga también pueden ser signos tempranos debido a la anemia resultante de la enfermedad de células falciformes. [22]
Aproximadamente la mitad de los pacientes con SCD autosómica recesiva clásica no experimentan una crisis hasta los cinco años de edad o más. [22] Algunos de estos pacientes, y otros que tienen otros genotipos, pueden presentar síntomas solo en condiciones de hipoxia moderada a severa y, por lo tanto, pueden no saber que tienen la enfermedad hasta más tarde en la vida. [16] Entre los niños mayores de 2 años, la presentación inicial más frecuente es un evento doloroso de tipo generalizado o variable, y una versión ligeramente menos común aparece como un evento de dolor torácico agudo. La dactilitis rara vez o nunca ocurre en niños mayores de 2 años. [22]
Los términos "crisis de células falciformes" o "crisis de drepanocitosis" pueden utilizarse para describir varias afecciones agudas independientes que se producen en pacientes con anemia falciforme y que dan lugar a anemia y crisis que pueden ser de muchos tipos, incluidas la crisis vasooclusiva , la crisis aplásica , la crisis de secuestro esplénico , la crisis hemolítica y otras. Los primeros síntomas [23] son un color amarillento de la piel o del blanco de los ojos que aparece cuando una gran cantidad de glóbulos rojos sufren hemólisis. La mayoría de los episodios de crisis de células falciformes duran entre cinco y siete días. [24] "Aunque la infección, la deshidratación y la acidosis (todas las cuales favorecen la drepanocitosis) pueden actuar como desencadenantes, en la mayoría de los casos no se identifica ninguna causa predisponente". [25]
La crisis vasooclusiva es causada por glóbulos rojos en forma de hoz que obstruyen los capilares y restringen el flujo sanguíneo a un órgano, lo que resulta en isquemia , dolor , necrosis y, a menudo, daño orgánico. La frecuencia, gravedad y duración de estas crisis varían considerablemente. Las crisis dolorosas se tratan con hidratación, analgésicos y transfusión de sangre ; el manejo del dolor requiere la administración de fármacos opioides a intervalos regulares hasta que la crisis se haya resuelto. Para las crisis más leves, un subgrupo de pacientes se maneja con medicamentos antiinflamatorios no esteroides como diclofenaco o naproxeno . Para las crisis más graves, la mayoría de los pacientes requieren tratamiento hospitalario para opioides intravenosos; los dispositivos de analgesia controlados por el paciente se utilizan comúnmente en este entorno. La crisis vasooclusiva que afecta a órganos como el pene [26] o los pulmones se considera una emergencia y se trata con transfusiones de glóbulos rojos. Se recomienda la espirometría incentiva , una técnica para estimular la respiración profunda para minimizar el desarrollo de atelectasia . [27]
El bazo se ve afectado con frecuencia en la enfermedad de células falciformes, ya que los glóbulos rojos en forma de hoz provocan el estrechamiento de los vasos sanguíneos y reducen la función para eliminar las células defectuosas. [28] Por lo general, se infarta antes del final de la infancia en personas con anemia de células falciformes. Este daño al bazo aumenta el riesgo de infección por organismos encapsulados ; [29] [30] se recomiendan antibióticos preventivos y vacunas para aquellos que carecen de una función adecuada del bazo .
Las crisis de secuestro esplénico son agrandamientos agudos y dolorosos del bazo, causados por el atrapamiento intraesplénico de glóbulos rojos y que resultan en una caída precipitada de los niveles de hemoglobina con el potencial de shock hipovolémico . Las crisis de secuestro se consideran una emergencia. Si no se tratan, los pacientes pueden morir en un plazo de 1 a 2 horas debido a una insuficiencia circulatoria. El tratamiento es de apoyo, a veces con transfusión de sangre. [31] Estas crisis son transitorias; continúan durante 3 a 4 horas y pueden durar un día. [32]
El síndrome torácico agudo se define por al menos dos de estos signos o síntomas: dolor torácico, fiebre, infiltrado pulmonar o anomalía focal, síntomas respiratorios o hipoxemia. [27] Es la segunda complicación más común y representa aproximadamente el 25% de las muertes en pacientes con ECF. La mayoría de los casos se presentan con crisis vasooclusivas y luego desarrollan síndrome torácico agudo. [33] [34] Sin embargo, aproximadamente el 80% de las personas tienen crisis vasooclusivas durante el síndrome torácico agudo. [35]
Las crisis aplásicas son casos de empeoramiento agudo de la anemia de base del paciente, que produce palidez , frecuencia cardíaca rápida y fatiga. Esta crisis normalmente es desencadenada por el parvovirus B19 , que afecta directamente la producción de glóbulos rojos al invadir los precursores de los glóbulos rojos y multiplicarse en ellos y destruirlos. [36] La infección por parvovirus [37] impide casi por completo la producción de glóbulos rojos durante dos o tres días. En individuos normales, esto tiene poca importancia, pero la vida acortada de los glóbulos rojos de los pacientes con anemia falciforme da lugar a una situación abrupta y potencialmente mortal. Los recuentos de reticulocitos caen drásticamente durante la enfermedad (lo que provoca reticulocitopenia ) y la rápida renovación de los glóbulos rojos conduce a la caída de la hemoglobina. Esta crisis tarda entre cuatro y siete días en desaparecer. La mayoría de los pacientes pueden tratarse con medidas de apoyo; algunos necesitan una transfusión de sangre. [38]
Las crisis hemolíticas son caídas aceleradas y agudas del nivel de hemoglobina. Los glóbulos rojos se descomponen a un ritmo más rápido. Esto es particularmente común en personas con deficiencia coexistente de G6PD . [39] Otra influencia de las crisis hemolíticas en la enfermedad de células falciformes es el estrés oxidativo en los eritrocitos, leucocitos y plaquetas. Cuando no hay suficiente producción de glóbulos rojos en la médula ósea, el oxígeno que el cuerpo recibe, procesa y transporta está desequilibrado con los antioxidantes del cuerpo. Hay un desequilibrio en las especies reactivas de oxígeno en las células, lo que conduce a una mayor producción de glóbulos rojos que no están adecuadamente oxigenados o formados. El estrés oxidativo puede conducir a anemia debido al desequilibrio de oxígeno en el tejido. [40]
El tratamiento es de apoyo, a veces con transfusiones de sangre. [27]
Una de las manifestaciones clínicas más tempranas es la dactilitis , que se presenta a los seis meses de edad y puede ocurrir en niños con rasgo drepanocítico. [41] La crisis puede durar hasta un mes. [42] Dado que la neumonía y la drepanocitosis en el pulmón pueden producir síntomas de síndrome torácico agudo, el paciente recibe tratamiento para ambas afecciones. [43] Puede desencadenarse por una crisis dolorosa, una infección respiratoria, una embolización de la médula ósea o posiblemente por atelectasia, administración de opiáceos o cirugía. [44] También pueden aparecer úlceras hematopoyéticas . [45]
La anemia de células falciformes puede provocar diversas complicaciones, entre ellas:




Normalmente, los seres humanos tienen en sus cuerpos hemoglobina A , que consta de dos cadenas alfa y dos cadenas beta, hemoglobina A2 , que consta de dos cadenas alfa y dos cadenas delta, y hemoglobina F (HbF), que consta de dos cadenas alfa y dos cadenas gamma. De estos tres tipos, la hemoglobina F domina hasta aproximadamente las 6 semanas de edad. Posteriormente, la hemoglobina A domina durante toda la vida. [63] En las personas diagnosticadas con anemia falciforme, al menos una de las subunidades de β-globina en la hemoglobina A se reemplaza por lo que se conoce como hemoglobina S. En la anemia falciforme, una forma común de anemia falciforme, la hemoglobina S reemplaza ambas subunidades de β-globina en la hemoglobina. [20]
La anemia falciforme tiene un patrón autosómico recesivo de herencia de los padres. [64] Los tipos de hemoglobina que una persona produce en los glóbulos rojos dependen de los genes de hemoglobina que hereda de sus padres. Si uno de los padres tiene anemia falciforme y el otro tiene el rasgo falciforme, entonces cualquier niño tiene un 50% de probabilidades de tener anemia falciforme y un 50% de probabilidades de tener el rasgo falciforme. Cuando ambos padres tienen el rasgo falciforme, cualquier niño tiene un 25% de probabilidades de tener anemia falciforme; un 25% de probabilidades de no tener alelos falciformes y un 50% de probabilidades de tener la condición heterocigótica. [65] [66] [ ¿ Fuente médica poco confiable? ]
La mutación del gen de la anemia falciforme probablemente surgió espontáneamente en diferentes áreas geográficas, [67] como lo sugiere el análisis de endonucleasas de restricción. Estas variantes se conocen como Camerún, Senegal, Benín, Bantú y Arabia Saudita-Asiática. Su importancia clínica se debe a que algunas están asociadas con niveles más altos de HbF, por ejemplo, las variantes de Senegal y Arabia Saudita-Asiática, y tienden a causar una enfermedad más leve. [68]
El defecto genético es una mutación de un solo nucleótido (ver polimorfismo de un solo nucleótido – SNP) ( el codón GAG cambia a GTG) del gen de la β-globina, que da como resultado que el glutamato (E/Glu) sea sustituido por valina (V/Val) en la posición 6 (sustitución E6V). [69] [nota 1] La hemoglobina S con esta mutación se conoce como HbS, a diferencia de la HbA adulta normal. Esta es normalmente una mutación benigna, que no causa efectos aparentes en las estructuras secundaria , terciaria o cuaternaria de la hemoglobina en condiciones de concentración normal de oxígeno . Sin embargo, bajo baja concentración de oxígeno , la HbS se polimeriza y forma precipitados fibrosos porque la forma desoxi de la hemoglobina expone un parche hidrofóbico en la proteína entre las hélices E y F (Phe 85, Leu 88). [70]
En las personas heterocigotas para la HbS ( portadoras de hemoglobina falciforme), los problemas de polimerización son menores porque el alelo normal puede producir la mitad de la hemoglobina. En las personas homocigotas para la HbS, la presencia de polímeros de cadena larga de HbS distorsiona la forma del glóbulo rojo, que pasa de ser liso, como una rosquilla , a ser irregular y lleno de púas, lo que lo hace frágil y susceptible de romperse dentro de los capilares . Los portadores tienen síntomas solo si se les priva de oxígeno (por ejemplo, al escalar una montaña) o cuando están gravemente deshidratados . [ cita requerida ]

El alelo responsable de la anemia de células falciformes se encuentra en el brazo corto del cromosoma 11 , más específicamente en el 11p15.5. Una persona que recibe el gen defectuoso tanto del padre como de la madre desarrolla la enfermedad; una persona que recibe un alelo defectuoso y uno sano permanece sana, pero puede transmitir la enfermedad y se la conoce como portadora o heterocigota. Los heterocigotos aún pueden contraer malaria, pero sus síntomas son generalmente menos graves. [71]
Debido a la ventaja adaptativa del heterocigoto, la enfermedad aún prevalece, especialmente entre personas con ascendencia reciente en áreas afectadas por la malaria, como África , el Mediterráneo , India y Medio Oriente . [72] La malaria fue históricamente endémica del sur de Europa, pero fue declarada erradicada a mediados del siglo XX, con la excepción de raros casos esporádicos. [73]
El parásito de la malaria tiene un ciclo de vida complejo y pasa parte de él en los glóbulos rojos. En un portador, la presencia del parásito de la malaria hace que los glóbulos rojos con hemoglobina defectuosa se rompan prematuramente, lo que hace que el parásito Plasmodium sea incapaz de reproducirse. Además, la polimerización de la Hb afecta la capacidad del parásito para digerir la Hb en primer lugar. Por lo tanto, en las zonas donde la malaria es un problema, las posibilidades de supervivencia de las personas en realidad aumentan si son portadoras de rasgos de anemia falciforme (selección para el heterocigoto). [ cita requerida ]
En los Estados Unidos, donde no hay malaria endémica, la prevalencia de la anemia de células falciformes entre las personas de ascendencia africana es menor (alrededor del 0,25%) que entre las personas de África occidental (alrededor del 4,0%) y está disminuyendo. Sin malaria endémica, la mutación de la anemia de células falciformes es puramente desventajosa y tiende a disminuir en la población afectada por selección natural y ahora artificialmente a través del cribado genético prenatal . Sin embargo, la comunidad afroamericana desciende de varios grupos étnicos africanos y no africanos, incluidos los esclavos estadounidenses. Por lo tanto, un grado de dilución genética a través del mestizaje con personas no africanas y una alta presión selectiva por la salud a través de la esclavitud (especialmente la trata de esclavos y el frecuentemente mortal Paso Medio ) pueden ser las explicaciones más plausibles para la menor prevalencia de la anemia de células falciformes (y, posiblemente, otras enfermedades genéticas) entre los afroamericanos en comparación con los africanos occidentales. Otro factor que limita la propagación de los genes de la anemia de células falciformes en América del Norte es la relativa ausencia de poligamia . En las sociedades polígamas, los varones afectados pueden tener muchos hijos con múltiples parejas. [74]
La pérdida de elasticidad de los glóbulos rojos es fundamental para la fisiopatología de la enfermedad de células falciformes. Los glóbulos rojos normales son bastante elásticos y tienen una forma de disco bicóncavo, lo que permite que las células se deformen para pasar a través de los capilares. [75] En la enfermedad de células falciformes, la baja tensión de oxígeno promueve la drepanocitosis de los glóbulos rojos y los episodios repetidos de drepanocitosis dañan la membrana celular y disminuyen la elasticidad de la célula. Estas células no vuelven a su forma normal cuando se restablece la tensión de oxígeno normal. Como consecuencia, estas células sanguíneas rígidas no pueden deformarse al pasar a través de capilares estrechos, lo que conduce a la oclusión de los vasos y a la isquemia . [ cita requerida ]
La anemia propiamente dicha de la enfermedad se debe a la hemólisis , la destrucción de los glóbulos rojos, debido a su forma. Aunque la médula ósea intenta compensar esto creando nuevos glóbulos rojos, no alcanza el ritmo de destrucción. [76] Los glóbulos rojos sanos suelen funcionar durante 90 a 120 días, pero los drepanocitos sólo duran entre 10 y 20 días. [77]
En la HbS, el hemograma completo revela niveles de hemoglobina en el rango de 6-8 g/dl con un alto recuento de reticulocitos (ya que la médula ósea compensa la destrucción de las células falciformes produciendo más glóbulos rojos). En otras formas de enfermedad de células falciformes, los niveles de Hb tienden a ser más altos. Un frotis de sangre puede mostrar características de hipoesplenismo ( células diana y cuerpos de Howell-Jolly ). [78]
La drepanocitosis de los glóbulos rojos en un frotis de sangre se puede inducir mediante la adición de metabisulfito de sodio . La presencia de hemoglobina drepanocítica también se puede demostrar con la "prueba de solubilidad drepanocítica" (también llamada "sickledex"). [79] Una mezcla de hemoglobina S (HbS) en una solución reductora (como el ditionito de sodio ) da un aspecto turbio, mientras que la Hb normal da una solución clara. [80]
Las formas anormales de hemoglobina se pueden detectar mediante electroforesis de hemoglobina , una forma de electroforesis en gel en la que los distintos tipos de hemoglobina se mueven a distintas velocidades. La hemoglobina falciforme (HgbS) y la hemoglobina C con drepanocitosis (HgbSC), las dos formas más comunes, se pueden identificar a partir de ahí. El diagnóstico se puede confirmar con cromatografía líquida de alto rendimiento . Rara vez se realizan pruebas genéticas , ya que otras investigaciones son altamente específicas para HbS y HbC. [81]
Una crisis aguda de anemia falciforme suele ser desencadenada por una infección. Por lo tanto, se debe realizar de forma sistemática un análisis de orina para detectar una infección oculta del tracto urinario y una radiografía de tórax para buscar una neumonía oculta. [82]
Las personas que son portadoras conocidas de la enfermedad o que corren el riesgo de tener un hijo con anemia de células falciformes pueden someterse a asesoramiento genético . Los asesores genéticos trabajan con las familias para discutir los beneficios, las limitaciones y la logística de las opciones de pruebas genéticas, así como el posible impacto de las pruebas y los resultados de las pruebas en el individuo. [83] [84] Durante el embarazo, las pruebas genéticas se pueden realizar en una muestra de sangre del feto o en una muestra de líquido amniótico . Durante el primer trimestre del embarazo, la muestra de vellosidades coriónicas (CVS) también es una técnica utilizada para el diagnóstico prenatal de SCD. [85] Dado que tomar una muestra de sangre de un feto tiene mayores riesgos, generalmente se utiliza esta última prueba. El cribado neonatal, a veces denominado cribado del recién nacido , no solo proporciona un método de detección temprana para las personas con enfermedad de células falciformes, sino que también permite la identificación de los grupos de personas que son portadoras del rasgo de células falciformes. [86] Los asesores genéticos pueden ayudar a las personas de color y a sus familias a abordar las disparidades raciales y étnicas que existen en la atención médica. [87]
En 2010, en los EE. UU. se debatió y se consideró de manera significativa la necesidad de realizar pruebas exhaustivas para detectar la anemia falciforme en los atletas. [88] [89] [90] [91] En 2012, la Sociedad Estadounidense de Hematología concluyó que no respaldaba la realización de pruebas o la divulgación del estado de anemia falciforme como requisito previo para participar en actividades deportivas debido a la falta de evidencia científica, la inconsistencia con las buenas prácticas médicas y la inconsistencia con la ética de la salud pública. Recomendó intervenciones universales para reducir las lesiones y muertes relacionadas con el esfuerzo que fueran efectivas para todos los atletas independientemente de su estado de anemia falciforme. [92]
El tratamiento implica una serie de medidas. Si bien históricamente se ha recomendado que las personas con anemia falciforme eviten el ejercicio, el ejercicio regular puede ser beneficioso para las personas. [93] Se debe evitar la deshidratación. [94] Se recomienda una dieta rica en calcio [95], pero la eficacia de los suplementos de vitamina D sigue siendo incierta. [96] La FDA respaldó el uso de L-glutamina a partir de los cinco años, ya que disminuye las complicaciones. [97]
Desde el nacimiento hasta los cinco años de edad, se recomienda administrar penicilina diariamente, debido al sistema inmunológico inmaduro que los hace más propensos a las enfermedades de la primera infancia. [98] La OMS había recomendado anteriormente la suplementación dietética de ácido fólico . [5] Una revisión Cochrane de 2016 sobre su uso encontró que "el efecto de la suplementación sobre la anemia y cualquier síntoma de anemia sigue sin estar claro" debido a la falta de evidencia médica. [99]

El efecto protector del rasgo drepanocítico no se aplica a las personas con enfermedad de células falciformes; de hecho, son más vulnerables a la malaria, ya que la causa más común de crisis dolorosas en los países palúdicos es la infección por malaria. Las personas con enfermedad de células falciformes que viven en países palúdicos deben recibir medicación de por vida para la prevención . [100]
La mayoría de las personas con enfermedad de células falciformes tienen episodios intensamente dolorosos llamados crisis vasooclusivas. Sin embargo, la frecuencia, gravedad y duración de estas crisis varían enormemente. Las crisis dolorosas se tratan sintomáticamente con analgésicos ; el manejo del dolor requiere la administración de fármacos opioides a intervalos regulares hasta que la crisis se haya resuelto. Para las crisis más leves, un subgrupo de pacientes se maneja con AINE (como diclofenaco o naproxeno ). Para las crisis más graves, la mayoría de los pacientes requieren tratamiento hospitalario para opioides intravenosos. [101]
Los líquidos adicionales, administrados por vía oral o intravenosa, son una parte rutinaria del tratamiento de las crisis vasooclusivas, pero la evidencia sobre la vía, la cantidad y el tipo de reemplazo de líquidos más efectivos sigue siendo incierta. [102]
En 2019, Crizanlizumab , un anticuerpo monoclonal dirigido contra la p-selectina , fue aprobado en los Estados Unidos para reducir la frecuencia de crisis vasooclusivas en personas de 16 años o más. [103]
La ecografía Doppler transcraneal (TCD) puede detectar a niños con anemia falciforme que tienen un alto riesgo de sufrir un accidente cerebrovascular. La prueba de ultrasonido detecta los vasos sanguíneos parcialmente obstruidos por la anemia falciforme midiendo la velocidad de la sangre que ingresa al cerebro, ya que la velocidad del flujo sanguíneo está inversamente relacionada con el diámetro arterial y, en consecuencia, la alta velocidad del flujo sanguíneo se correlaciona con el estrechamiento de las arterias. [104] En 2002, el Instituto Nacional de Salud (NIH) emitió una declaración recomendando que los niños con anemia falciforme se sometan a la ecografía Doppler transcraneal anualmente, y en 2014, un panel de expertos convocado por el NIH emitió pautas que reiteraban la misma recomendación. Una revisión de los registros médicos, realizada por la hematóloga Dra. Julie Kanter en la Universidad de Alabama en Birmingham, mostró que, en promedio, solo el 48,4 por ciento de los niños con anemia falciforme se someten a la prueba de ecografía recomendada. [105]
Un estudio de los NIH de 1994 mostró que los niños con riesgo de sufrir accidentes cerebrovasculares que recibieron transfusiones de sangre tuvieron una tasa anual de accidentes cerebrovasculares de menos del 1 por ciento, mientras que aquellos niños que no recibieron transfusiones de sangre tuvieron una tasa de accidentes cerebrovasculares del 10 por ciento por año. (Véase también el estudio de 1998 en el New England Journal of Medicine . [104] ) Además de las ecografías y las transfusiones de sangre, el fármaco genérico económico hidroxiurea puede reducir el riesgo de daño irreversible a los órganos y al cerebro. Las directrices de los NIH publicadas en 2014 establecen que todos los niños y adolescentes deben tomar hidroxiurea, al igual que los adultos con complicaciones graves o tres o más crisis de dolor en un año. [105]
El tratamiento es similar al de una crisis vasooclusiva, con la adición de antibióticos (generalmente una quinolona o un macrólido, ya que se cree que las bacterias deficientes en la pared celular ["atípicas"] contribuyen al síndrome), [106] suplementos de oxígeno para la hipoxia y observación estrecha. En ausencia de evidencia de alta calidad con respecto a la efectividad de los antibióticos para el síndrome torácico agudo en personas con enfermedad de células falciformes, no existe un tratamiento antibiótico estándar a partir de 2019. [107] Se recomienda que las personas con sospecha de síndrome torácico agudo sean ingresadas en el hospital con empeoramiento del gradiente Aa como indicación de ingreso en la UCI. [27]
Si el infiltrado pulmonar empeora o aumentan los requerimientos de oxígeno, está indicada una transfusión sanguínea simple o una exanguinotransfusión . Esta última implica el intercambio de una porción significativa de la masa de glóbulos rojos de la persona por glóbulos rojos normales, lo que disminuye el nivel de hemoglobina S en la sangre del paciente. Sin embargo, actualmente hay evidencia incierta sobre los posibles beneficios o daños de la transfusión sanguínea para el síndrome torácico agudo en personas con enfermedad de células falciformes. [108]
La hidroxiurea , también conocida como hidroxicarbamida , probablemente reduce la frecuencia de episodios dolorosos y el riesgo de enfermedad potencialmente mortal o muerte, pero actualmente no hay evidencia suficiente con respecto al riesgo de efectos adversos. [109] La hidroxiurea y la flebotomía combinadas pueden ser más efectivas que la transfusión y la quelación combinadas en términos de dolor, enfermedad potencialmente mortal y riesgo de muerte. [109]
Fue el primer fármaco aprobado para el tratamiento de la anemia de células falciformes, y se demostró que reducía el número y la gravedad de los ataques en 1995 [110] y que posiblemente aumentaba el tiempo de supervivencia en un estudio de 2003. [111] Esto se logra, en parte, reactivando la producción de hemoglobina fetal en lugar de la hemoglobina S que causa la anemia de células falciformes. La hidroxiurea se había utilizado anteriormente como agente de quimioterapia , y existe cierta preocupación de que su uso a largo plazo pueda ser perjudicial, pero este riesgo es inexistente o muy pequeño y los beneficios probablemente superen los riesgos. [21] [112]
Voxelotor fue aprobado en los Estados Unidos en 2019 para aumentar la hemoglobina en personas con enfermedad SS. [113]
Las transfusiones de sangre se utilizan a menudo en el tratamiento de la enfermedad de células falciformes en casos agudos y para prevenir complicaciones al disminuir la cantidad de glóbulos rojos (RBC) que pueden formar células falciformes al agregar glóbulos rojos normales. [114] En los niños, se ha demostrado que la terapia de transfusión de glóbulos rojos preventiva reduce el riesgo de un primer accidente cerebrovascular o un accidente cerebrovascular silencioso cuando la ecografía Doppler transcraneal muestra un flujo sanguíneo cerebral anormal. [6] En aquellos que han sufrido un accidente cerebrovascular previo, también reduce el riesgo de accidente cerebrovascular recurrente y accidentes cerebrovasculares silenciosos adicionales. [115] [116]
Los trasplantes de médula ósea han demostrado ser eficaces en niños; son la única cura conocida para la anemia drepanocítica. [117] Sin embargo, los trasplantes de médula ósea son difíciles de obtener debido a la tipificación específica de HLA necesaria. Lo ideal sería que un pariente cercano (alogénico) donara la médula ósea necesaria para el trasplante. El pariente cercano debe tener el mismo tipo de sangre que el paciente. Se están desarrollando algunas terapias genéticas que alterarían las células madre de la médula ósea del propio paciente ex vivo, que luego se pueden trasplantar nuevamente al paciente después de que la quimioterapia elimine las células originales no modificadas. [118]
Trasplante de células madre hematopoyéticas (TCMH)
El tratamiento de la anemia de células falciformes implica implícitamente el trasplante de células madre hematopoyéticas (TCMH). [119] Esto implica reemplazar las células madre disfuncionales en el caso con huesos sanos de un donante compatible. Encontrar al cliente ideal, por lo general un donante de reserva o alguien casi compatible, es esencial para el éxito del proceso. Sin embargo, todavía existen algunos inconvenientes para el trasplante de células madre hematopoyéticas (TCMH), como el requisito de detalles precisos para inhibir el sistema susceptible y problemas de que las nuevas células no se arraiguen. Sin embargo, un TCMH exitoso puede resultar en una cura a largo plazo de la anemia de células falciformes para el paciente. [120]
En el tratamiento de la necrosis avascular del hueso en personas con enfermedad de células falciformes, el objetivo del tratamiento es reducir o detener el dolor y mantener la movilidad de las articulaciones . [50] Las opciones de tratamiento actuales incluyen el reposo de la articulación, la fisioterapia , los analgésicos , la cirugía de reemplazo articular o el injerto óseo . [50] Se necesitan ensayos controlados, aleatorizados y de alta calidad para evaluar la opción de tratamiento más eficaz y determinar si una combinación de fisioterapia y cirugía es más eficaz que la fisioterapia sola. [121] [122]
Las terapias psicológicas como la educación del paciente , la terapia cognitiva , la terapia conductual y la psicoterapia psicodinámica , que tienen como objetivo complementar los tratamientos médicos actuales, requieren más investigaciones para determinar su eficacia. [28]
En 2023, tanto el exagamglogene autotemcel como el lovotibeglogene autotemcel fueron aprobados para el tratamiento de la enfermedad de células falciformes. [13] [123]
Alrededor del 90% de las personas sobreviven hasta los 20 años, y cerca del 50% sobreviven más allá de los 50 años . [124] En 2001, según un estudio realizado en Jamaica, la supervivencia media estimada para las personas con anemia drepanocítica homocigótica era de 53 años para los hombres y de 58 años para las mujeres. [125] Se desconoce la esperanza de vida en gran parte del mundo en desarrollo. [126] En 1975, alrededor del 7,3% de las personas con anemia drepanocítica morían antes de cumplir 23 años; mientras que en 1989, el 2,6% de las personas con anemia drepanocítica morían antes de cumplir 20 años. [127] : 348
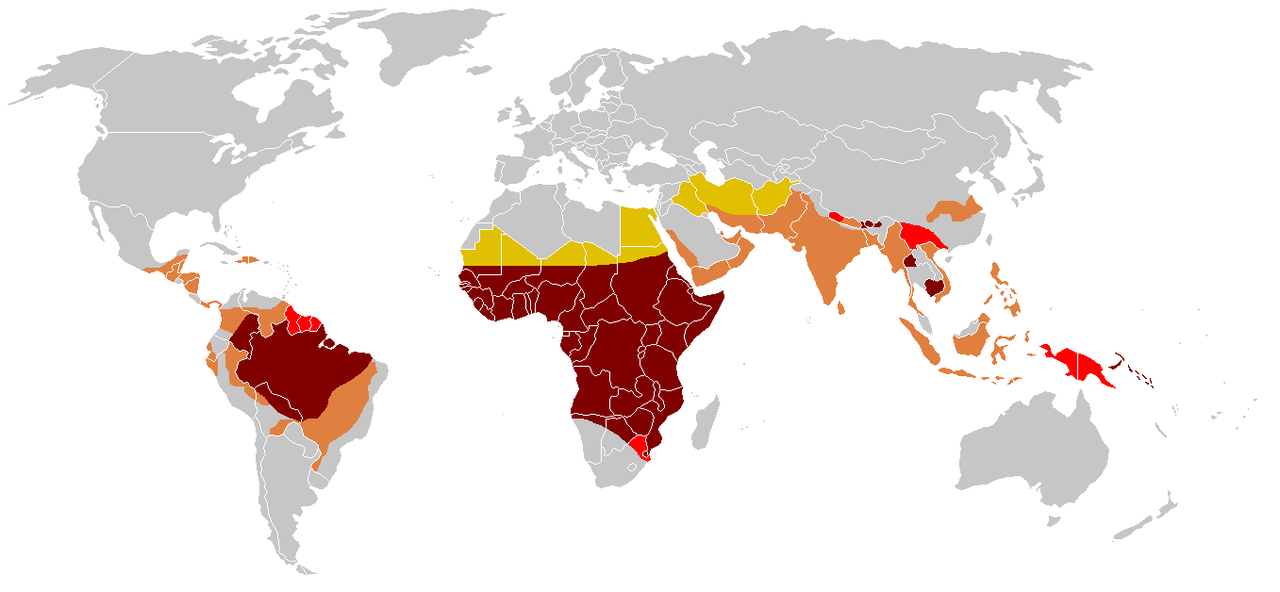
El gen HbS se puede encontrar en todos los grupos étnicos. [128] La frecuencia más alta de la enfermedad de células falciformes se encuentra en las regiones tropicales, particularmente en África subsahariana, las regiones tribales de la India y Oriente Medio. [129] Se cree que alrededor del 80% de los casos de enfermedad de células falciformes ocurren en África subsahariana . [16] La migración de poblaciones sustanciales de estas áreas de alta prevalencia a países de baja prevalencia en Europa ha aumentado drásticamente en las últimas décadas y en algunos países europeos, la enfermedad de células falciformes ahora ha superado condiciones genéticas más familiares como la hemofilia y la fibrosis quística . [130] En 2015, resultó en alrededor de 114.800 muertes. [8]
La enfermedad de células falciformes se presenta con mayor frecuencia entre personas cuyos antepasados vivieron en regiones subsaharianas tropicales y subtropicales donde la malaria es o fue común. En los lugares donde la malaria es común, ser portador de un solo alelo (rasgo) de la enfermedad de células falciformes confiere una ventaja heterocigótica ; los seres humanos con uno de los dos alelos de la enfermedad de células falciformes muestran síntomas menos graves cuando se infectan con malaria. [131]
Esta afección se hereda con un patrón autosómico recesivo, lo que significa que ambas copias del gen en cada célula tienen mutaciones. Cada uno de los padres es portador de una copia del gen mutado, pero por lo general no presentan signos ni síntomas de la afección. [132]
Tres cuartas partes de los casos de anemia falciforme se producen en África. Un informe reciente de la OMS estimó que alrededor del 2% de los recién nacidos en Nigeria estaban afectados por anemia falciforme, lo que da un total de 150.000 niños afectados nacidos cada año solo en Nigeria. La frecuencia de portadores varía entre el 10 y el 40% en África ecuatorial, disminuyendo al 1-2% en la costa norteafricana y a menos del 1% en Sudáfrica. [133] Los estudios en África muestran una disminución significativa en la tasa de mortalidad infantil, de 2 a 16 meses de edad, debido al rasgo falciforme. Esto sucedió en áreas de casos predominantes de malaria. [134]
Uganda tiene la quinta mayor carga de enfermedad de células falciformes en África. [135] Un estudio indica que 20 000 bebés por año nacen con enfermedad de células falciformes, con el rasgo de células falciformes en un 13,3% y con la enfermedad en un 0,7%. [136]
El número de personas con la enfermedad en los Estados Unidos es de aproximadamente 100.000 (una de cada 3.300), y afecta principalmente a estadounidenses de ascendencia africana subsahariana. [137] En los Estados Unidos, aproximadamente uno de cada 365 niños afroamericanos y uno de cada 16.300 niños hispanoamericanos tienen anemia de células falciformes. [138] La esperanza de vida de los hombres con anemia falciforme es de aproximadamente 42 años, mientras que las mujeres viven aproximadamente seis años más. [139] Otros 2 millones son portadores del rasgo de células falciformes. [140] La mayoría de los bebés con anemia falciforme nacidos en los Estados Unidos se identifican mediante pruebas de detección neonatal de rutina. A partir de 2016, los 50 estados incluyen la detección de la enfermedad de células falciformes como parte de su detección neonatal. [141] Se toma una muestra de sangre del recién nacido a través de un pinchazo en el talón y se envía a un laboratorio para su análisis. El bebé debe haber estado comiendo durante un mínimo de 24 horas antes de que se pueda realizar la prueba del talón. Algunos estados también exigen que se realice un segundo análisis de sangre cuando el bebé tenga dos semanas de vida para garantizar los resultados. [142]
La anemia de células falciformes es el trastorno genético más común entre los afroamericanos. Aproximadamente el 8% son portadores y 1 de cada 375 nace con la enfermedad. [143] Los defensores de los pacientes de la enfermedad de células falciformes se han quejado de que recibe menos fondos gubernamentales y privados para la investigación que enfermedades raras similares, como la fibrosis quística , y el investigador Elliott Vichinsky dice que esto muestra discriminación racial o el papel de la riqueza en la defensa de la atención médica. [144] En general, sin considerar la raza, aproximadamente el 1,5% de los bebés nacidos en los Estados Unidos son portadores de al menos una copia del gen mutante (causante de la enfermedad). [145]

Como resultado del crecimiento de la población en las regiones africanas y caribeñas de ultramar de Francia y la inmigración desde el norte de África y África subsahariana a Francia continental, la enfermedad de células falciformes se ha convertido en un importante problema de salud en Francia. [146] La SCD se ha convertido en la enfermedad genética más común en el país, con una prevalencia general al nacer de uno en 2415 en Francia metropolitana , por delante de la fenilcetonuria (uno en 10 862), el hipotiroidismo congénito (uno en 3132), la hiperplasia suprarrenal congénita (uno en 19 008) y la fibrosis quística (uno en 5014) para el mismo período de referencia. [147]

Desde el año 2000, se ha realizado un cribado neonatal de la anemia drepanocítica a nivel nacional para todos los recién nacidos definidos como "en riesgo" de padecerla según su origen étnico (definidos como aquellos nacidos de padres originarios de África subsahariana, el norte de África, la zona mediterránea (sur de Italia, Grecia y Turquía ), la península arábiga, las islas francesas de ultramar y el subcontinente indio). [148]
En el Reino Unido, se cree que entre 12.000 y 15.000 personas padecen la enfermedad de células falciformes [149] y que tan solo en Inglaterra hay unos 250.000 portadores de la enfermedad. Como el número de portadores es solo una estimación, a todos los recién nacidos del Reino Unido se les realiza un análisis de sangre de rutina para detectar la enfermedad [150] . Debido a que muchos adultos de los grupos de alto riesgo no saben si son portadores, a las mujeres embarazadas y a ambos miembros de la pareja se les ofrece la posibilidad de hacerse una prueba de detección para que puedan recibir asesoramiento si tienen el rasgo de células falciformes [151] . Además, a los donantes de sangre de los grupos de alto riesgo también se les realiza una prueba de detección para confirmar si son portadores y si su sangre se filtra correctamente [152] . A los donantes que resultan ser portadores se les informa y su sangre, aunque a menudo se utiliza para personas del mismo grupo étnico, no se utiliza para personas con enfermedad de células falciformes que requieren una transfusión de sangre [153] .
En Arabia Saudita , aproximadamente el 4,2% de la población es portadora del gen de la anemia falciforme y el 0,26% tiene la enfermedad de células falciformes. La prevalencia más alta se encuentra en la provincia oriental, donde aproximadamente el 17% de la población es portadora del gen y el 1,2% tiene la enfermedad de células falciformes. [154] En 2005, Arabia Saudita introdujo una prueba prematrimonial obligatoria que incluía la electroforesis de HB, cuyo objetivo era reducir la incidencia de la anemia falciforme y la talasemia . [155]
En Bahréin , un estudio publicado en 1998 que abarcó a unas 56.000 personas en hospitales de Bahréin encontró que el 2% de los recién nacidos tienen anemia falciforme, el 18% de las personas encuestadas tienen el rasgo de anemia falciforme y el 24% eran portadores de la mutación genética que causa la enfermedad. [156] El país comenzó a realizar pruebas de detección a todas las mujeres embarazadas en 1992, y se empezó a realizar pruebas a los recién nacidos si la madre era portadora. En 2004, se aprobó una ley que exigía que las parejas que planeaban casarse se sometieran a asesoramiento prematrimonial gratuito . Estos programas fueron acompañados de campañas de educación pública. [157]
La enfermedad de células falciformes es común en algunos grupos étnicos de la India central, [158] donde la prevalencia ha oscilado entre el 9,4 y el 22,2% en las zonas endémicas de Madhya Pradesh , Rajasthan y Chhattisgarh . [159] También es endémica entre los tharu de Nepal y la India; sin embargo, tienen una tasa siete veces menor de malaria a pesar de vivir en una zona infestada de malaria. [160]
En Jamaica , el 10% de la población es portadora del gen de la anemia falciforme, lo que lo convierte en el trastorno genético más prevalente en el país. [161]
El primer informe moderno sobre la enfermedad de células falciformes puede haber sido en 1846, donde se discutió la autopsia de un esclavo fugitivo ejecutado; el hallazgo clave fue la ausencia del bazo. [162] [163] Según se informa, los esclavos africanos en los Estados Unidos mostraron resistencia a la malaria, pero eran propensos a las úlceras en las piernas. [163] Las características anormales de los glóbulos rojos, que más tarde prestaron su nombre a la enfermedad, fueron descritas por primera vez por Ernest E. Irons (1877-1959), interno del cardiólogo de Chicago y profesor de medicina James B. Herrick (1861-1954), en 1910. Irons vio células "peculiares alargadas y en forma de hoz" en la sangre de un hombre llamado Walter Clement Noel, un estudiante de odontología de primer año de 20 años de Granada. Noel había sido ingresado en el Hospital Presbiteriano de Chicago en diciembre de 1904 con anemia. [18] [164] Noel fue readmitido varias veces durante los siguientes tres años por " reumatismo muscular " y "ataques biliosos", pero completó sus estudios y regresó a la capital de Granada (St. George's) para ejercer la odontología . Murió de neumonía en 1916 y está enterrado en el cementerio católico de Sauteurs , en el norte de Granada. [18] [19] Poco después del informe de Herrick, apareció otro caso en el Virginia Medical Semi-Monthly con el mismo título, "Peculiar Elongated and Falke-Shaped Red Blood Corpuscles in a Case of Severe Anemia". [165] Este artículo se basa en un paciente ingresado en el Hospital de la Universidad de Virginia el 15 de noviembre de 1910. [166] En la descripción posterior de Verne Mason en 1922, se utiliza por primera vez el nombre "anemia de células falciformes". [19] [167] Los problemas infantiles relacionados con la enfermedad de células falciformes no se informaron hasta la década de 1930, a pesar del hecho de que esto no puede haber sido poco común en las poblaciones afroamericanas. [163]
El médico de Memphis Lemuel Diggs , un prolífico investigador de la enfermedad de células falciformes, introdujo por primera vez la distinción entre la enfermedad de células falciformes y el rasgo en 1933, aunque hasta 1949, las características genéticas no habían sido dilucidadas por James V. Neel y EA Beet. [19] 1949 fue el año en que Linus Pauling describió el comportamiento químico inusual de la hemoglobina S y lo atribuyó a una anomalía en la propia molécula. [19] [168] El cambio molecular en HbS fue descrito en 1956 por Vernon Ingram . [169] A finales de la década de 1940 y principios de la de 1950 se comprendió mejor el vínculo entre la malaria y la enfermedad de células falciformes. En 1954, la introducción de la electroforesis de hemoglobina permitió el descubrimiento de subtipos particulares, como la enfermedad HbSC. [19]
En los años 1970 y 1980 se introdujeron estudios de historia natural a gran escala y otros estudios de intervención, lo que condujo al uso generalizado de la profilaxis contra las infecciones neumocócicas, entre otras intervenciones. La película para televisión de Bill Cosby , ganadora de un premio Emmy en 1972, To All My Friends on Shore , describía la historia de los padres de un niño con anemia de células falciformes. [170] En los años 1990 se desarrolló la hidroxicarbamida y en 2007 aparecieron informes de curación mediante trasplante de médula ósea. [19]
Algunos textos antiguos se refieren a ella como drepanocitosis. [171]
La enfermedad de células falciformes se considera con frecuencia una discapacidad. [172] A partir del 15 de septiembre de 2017, la Administración del Seguro Social de los EE. UU. emitió una Resolución de Interpretación de Políticas que proporciona información de fondo sobre la enfermedad de células falciformes y una descripción de cómo el Seguro Social evalúa la enfermedad durante su proceso de adjudicación de reclamos por discapacidad. [173] [174]
En los EE. UU., existen estigmas en torno a la anemia drepanocítica que desalientan a las personas con anemia drepanocítica a recibir la atención necesaria. Estos estigmas afectan principalmente a las personas de ascendencia afroamericana y latinoamericana, según el Instituto Nacional del Corazón, los Pulmones y la Sangre. [175] Las personas con anemia drepanocítica experimentan el impacto de los estigmas de la enfermedad en múltiples aspectos de la vida, incluido el bienestar social y psicológico. Los estudios han demostrado que las personas con anemia drepanocítica con frecuencia sienten que deben mantener su diagnóstico en secreto para evitar la discriminación en el lugar de trabajo y también entre pares en las relaciones. [176] En la década de 1960, el gobierno de los EE. UU. apoyó iniciativas para la detección de enfermedades genéticas en el lugar de trabajo en un intento de proteger a las personas con anemia drepanocítica. Al realizar esta detección, se pretendía que los empleados no se encontraran en entornos que pudieran ser potencialmente dañinos y desencadenar la anemia drepanocítica. [177]
Uganda tiene la quinta carga más alta de enfermedad de células falciformes (ECF) en el mundo. [178] En Uganda , existe un estigma social para aquellos con enfermedad de células falciformes debido a la falta de conocimiento general de la enfermedad. La brecha general en el conocimiento sobre la enfermedad de células falciformes se nota entre los adolescentes y adultos jóvenes debido al secreto culturalmente sancionado sobre la enfermedad. [178] Si bien la mayoría de las personas han oído hablar en general de la enfermedad, una gran parte de la población está relativamente mal informada sobre cómo se diagnostica o se hereda la ECF. Aquellos que están informados sobre la enfermedad se enteraron de ella por familiares o amigos y no por profesionales de la salud . El hecho de no proporcionar al público información sobre la enfermedad de células falciformes da como resultado una población con una comprensión deficiente de las causas de la enfermedad, los síntomas y las técnicas de prevención. [179] Las diferencias, físicas y sociales, que surgen en las personas con enfermedad de células falciformes, como ictericia, retraso del crecimiento físico y retraso de la madurez sexual, también pueden llevarlos a convertirse en blancos de acoso, rechazo y estigma. [178]
Los datos recopilados sobre la enfermedad de células falciformes en Uganda no se han actualizado desde principios de la década de 1970. La deficiencia de datos se debe a la falta de fondos gubernamentales para la investigación, a pesar de que los ugandeses mueren diariamente por anemia de células falciformes. [180] Los datos muestran que la frecuencia del rasgo de la enfermedad de células falciformes es del 20% de la población de Uganda. [180] Esto significa que 66 millones de personas corren el riesgo de tener un hijo con enfermedad de células falciformes. [180] También se estima que alrededor de 25.000 ugandeses nacen cada año con anemia de células falciformes y el 80% de esas personas no viven más allá de los cinco años. [180] La anemia de células falciformes también contribuye en un 25% a la tasa de mortalidad infantil en Uganda. [180] El pueblo bamba de Uganda, ubicado en el suroeste del país, es portador del 45% del gen, que es la frecuencia de rasgo más alta registrada en el mundo. [180] La Clínica de Anemia Falciforme en Mulago es la única clínica de enfermedad de células falciformes en el país y atiende en promedio a 200 pacientes por día. [180]
El estigma en torno a la enfermedad es particularmente grave en las regiones del país que no están tan afectadas. Por ejemplo, los ugandeses del este tienden a tener más conocimientos sobre la enfermedad que los ugandeses del oeste, que son más propensos a creer que la anemia falciforme es el resultado de un castigo de Dios o de la brujería . [181] Otros conceptos erróneos sobre la anemia falciforme incluyen la creencia de que es causada por factores ambientales, pero, en realidad, la anemia falciforme es una enfermedad genética. [182] Se han hecho esfuerzos en toda Uganda para abordar los conceptos erróneos sociales sobre la enfermedad. En 2013, se creó la Uganda Sickle Cell Rescue Foundation para difundir la conciencia sobre la anemia falciforme y combatir el estigma social asociado a la enfermedad. [183] Además de los esfuerzos de esta organización, es necesario incluir la educación sobre la anemia falciforme en los programas de educación sanitaria comunitaria preexistentes con el fin de reducir la estigmatización de la anemia falciforme en Uganda. [179]
El estigma profundamente arraigado de la anemia de células falciformes en la sociedad hace que las familias a menudo oculten el estado de enfermedad de sus familiares por miedo a ser etiquetados, maldecidos o excluidos de los eventos sociales. [184] A veces en Uganda, cuando se confirma que un miembro de la familia tiene anemia de células falciformes, se evitan las relaciones íntimas con todos los miembros de la familia. [184] La estigmatización y el aislamiento social que tienden a experimentar las personas con anemia de células falciformes es a menudo la consecuencia de conceptos erróneos populares de que las personas con anemia de células falciformes no deben socializar con personas libres de la enfermedad. Esta mentalidad priva a las personas con anemia de células falciformes del derecho a participar libremente en actividades comunitarias como todos los demás. [178] El estigma relacionado con la anemia de células falciformes y el aislamiento social en las escuelas, especialmente, pueden hacer que la vida de los jóvenes que viven con anemia de células falciformes sea extremadamente difícil. [178] Para los niños en edad escolar que viven con anemia de células falciformes, el estigma al que se enfrentan puede conducir al rechazo de los compañeros. [178] El rechazo de los compañeros implica la exclusión de grupos o reuniones sociales. Esto a menudo lleva a que el individuo excluido experimente angustia emocional y puede dar como resultado un bajo rendimiento académico, el rechazo a la escuela y un fracaso laboral más adelante en la vida. [178] Este aislamiento social también es probable que afecte negativamente la autoestima y la calidad de vida general de las personas con ECF . [178]
Las madres de niños con anemia falciforme tienden a recibir cantidades desproporcionadas de estigma por parte de sus pares y miembros de la familia. A menudo se culpa a estas mujeres por el diagnóstico de anemia falciforme de su hijo, especialmente si la anemia falciforme no está presente en generaciones anteriores, debido a la sospecha de que la mala salud del niño puede haber sido causada por el fracaso de la madre para implementar medidas de salud preventivas o promover un entorno saludable para que su hijo prospere. [182] La dependencia de teorías relacionadas con factores ambientales para culpar a la madre refleja el escaso conocimiento de muchos ugandeses sobre cómo se adquiere la enfermedad, ya que está determinada por la genética, no por el medio ambiente. [182] Las madres de niños con anemia falciforme también suelen quedarse con recursos muy limitados para salvaguardar su futuro contra el estigma de tener anemia falciforme. [182] Esta falta de acceso a los recursos es resultado de sus roles subordinados dentro de las estructuras familiares, así como de las disparidades de clase que obstaculizan la capacidad de muchas madres para satisfacer los costos y responsabilidades adicionales del cuidado infantil. [182]
Las mujeres que viven con anemia falciforme y quedan embarazadas a menudo enfrentan una discriminación extrema y desánimo en Uganda. Estas mujeres son frecuentemente tildadas por sus pares de irresponsables por tener un bebé mientras viven con enfermedad de células falciformes o incluso tener relaciones sexuales mientras viven con anemia de células falciformes. Carga del rasgo y la enfermedad de células falciformes en el Estudio de Vigilancia de la Anemia de Células Falciformes de Uganda (US3): un estudio transversal Las críticas y los juicios que reciben estas mujeres, no solo de los profesionales de la salud sino también de sus familias, a menudo las dejan sintiéndose solas, deprimidas, ansiosas, avergonzadas y con muy poco apoyo social . Carga del rasgo y la enfermedad de células falciformes en el Estudio de Vigilancia de la Anemia de Células Falciformes de Uganda (US3): un estudio transversal - The Lancet Global Health La mayoría de las mujeres embarazadas con anemia de células falciformes también se convierten en madres solteras, ya que es común que sus parejas masculinas las abandonen y aleguen que no sabían que su pareja tenía anemia de células falciformes. Padres de niños con anemia de células falciformes mal preparados y mal informados: es hora de repensar las campañas de concienciación El abandono que experimentan estas mujeres no solo les causa angustia emocional, sino que este bajo nivel de apoyo parental puede estar relacionado con síntomas depresivos y, en general, menor calidad de vida para el niño una vez nacido. [185]
En 2021, se descubrió que muchos pacientes tenían miedo de visitar los hospitales, tal era el nivel de ignorancia entre el personal, por lo que compraron analgésicos para tratarse fuera del NHS. A menudo esperaban mucho tiempo para recibir alivio del dolor y, a veces, se sospechaba que tenían un comportamiento de "búsqueda de medicamentos". Los retrasos en el tratamiento, la falta de información al equipo de hematología del hospital y el mal manejo del dolor habían causado muertes. El personal de hematología especializado prefería trabajar en hospitales docentes más grandes, lo que provocó una escasez de experiencia en otros lugares. [186] En 2021, el NHS inició su primer tratamiento nuevo en 20 años para la anemia falciforme. Esto implicó el uso de Crizanlizumab , un medicamento que se administra mediante goteo transfusional, lo que reduce el número de visitas a Urgencias por parte de los pacientes. Se puede acceder al tratamiento, a través de consultores, en cualquiera de los diez nuevos centros establecidos en todo el país. [187] Sin embargo, ese mismo año, un grupo parlamentario multipartidario elaboró un informe sobre la anemia falciforme y la talasemia titulado "Nadie está escuchando". [188] En parte como respuesta a esto, el 19 de junio de 2022, Día Mundial de la Anemia Falciforme, el NHS lanzó una campaña llamada "¿Puedes decir que es anemia falciforme?". La campaña tenía un doble objetivo. Uno era aumentar la conciencia de los signos y síntomas clave de este trastorno sanguíneo para que las personas estuvieran tan alertas a los signos de una crisis de anemia falciforme como a un ataque cardíaco o un derrame cerebral inminente. El segundo objetivo era establecer un nuevo programa de capacitación para ayudar a los paramédicos, el personal de accidentes y emergencias, los cuidadores y el público en general a atender de manera efectiva a los pacientes en crisis. [189]
Si bien el trasplante de sangre del cordón umbilical puede curar potencialmente la enfermedad, solo el 10% de los pacientes pueden contar con un donante adecuado. [190] Alrededor del 7% de las personas también mueren como resultado del procedimiento y puede producirse una enfermedad de injerto contra huésped . [190]
Las enfermedades como la anemia falciforme, en las que el fenotipo o la función celular normal de una persona pueden restaurarse en las células que padecen la enfermedad mediante una copia normal del gen que está mutado, pueden ser buenas candidatas para el tratamiento con terapia génica. No se conocen los riesgos y beneficios relacionados con la terapia génica para la anemia falciforme. [191]
En 2001, se informó que la enfermedad de células falciformes había sido tratada con éxito en ratones mediante terapia génica . [192] [193] Los investigadores utilizaron un vector viral para hacer que los ratones, que tienen esencialmente el mismo defecto que causa la enfermedad de células falciformes humana, expresaran la producción de hemoglobina fetal (HbF), que un individuo normalmente deja de producir poco después del nacimiento. En humanos, se sabe que el uso de hidroxiurea para estimular la producción de HbF alivia temporalmente los síntomas de la enfermedad de células falciformes. Los investigadores demostraron que este método de terapia génica es una forma más permanente de aumentar la producción terapéutica de HbF. [194]
En 2014 se iniciaron los ensayos clínicos de fase 1 de terapia génica para la anemia falciforme en humanos. Los ensayos clínicos evaluaron la seguridad de la médula ósea modificada con vectores lentivirales para adultos con anemia falciforme grave. [195] [196] En marzo de 2017 se publicó un informe de caso de la primera persona tratada, y desde entonces se ha tratado a algunas personas más. [197] [198]
Las plataformas de edición genética como CRISPR/Cas9 se han utilizado para corregir la mutación causante de la enfermedad en células madre hematopoyéticas extraídas de una persona con la afección. [199] En julio de 2019, la herramienta de edición genética CRISPR se utilizó para editar células de la médula ósea de una persona con SCD para aumentar la hemoglobina fetal inhibiendo el gen BCL11A . [200] [201] Varios investigadores han considerado las implicaciones éticas de que la SCD sea una de las primeras aplicaciones potenciales de la tecnología CRISPR, dados los abusos históricos y el abandono de la comunidad afroamericana por parte del campo médico. [202]
En 2017, doce ensayos clínicos se centraron en la terapia génica para tratar la anemia de células falciformes. De esos 12 ensayos, cuatro de ellos reemplazaron el gen HBB mutado por uno sano. Tres ensayos utilizaron Mozobil, un medicamento utilizado para tratar tipos de cáncer, para determinar si el aumento de células madre se puede utilizar para la terapia génica. Un ensayo se centró en analizar muestras de médula ósea de pacientes con anemia de células falciformes. Otro ensayo experimentó con el uso de sangre del cordón umbilical de bebés con y sin anemia de células falciformes para desarrollar una terapia génica. [203]
En noviembre de 2023, los reguladores del Reino Unido aprobaron un tratamiento genético que utiliza la herramienta de edición genética CRISPR para el tratamiento de la anemia de células falciformes y también para el trastorno sanguíneo beta talasemia dependiente de transfusiones . [14] [123]
No existe evidencia médica sólida para determinar los riesgos y los beneficios potenciales relacionados con el tratamiento de personas con enfermedad de células falciformes con trasplantes de células madre hematopoyéticas. [204]
{{cite journal}}: CS1 maint: unfit URL (link)twice-daily prophylactic penicillin beginning in early infancy and continuing through at least age 5
... teams report that two strategies for directly fixing malfunctioning blood cells have dramatically improved the health of a handful of people with these genetic diseases.
{{cite journal}}: CS1 maint: DOI inactive as of June 2024 (link)